When I have data-crunching to do, I like to do it in the browser. Excel gives me a headache. Teaching myself how to write Excel macros doesn't leave me with a transferable skill (a skill that can be used outside of Excel); it just makes me an expert at Excel. Which I don't want to be.
At least if I crunch data with JavaScript, I learn JavaScript skills that I can apply to web-page design, AJAX programming, Scalable Vector Graphics programming, Canvas programming, and lots of other scenarios.
What do I mean by crunching data in the browser? Say you have a text file containing some data. Open the text file in Chrome or Firefox, then use Control-Shift-J to get to the browser's JavaScript console. There you are. You're ready to crunch data.
How? Let's say your text file contains a table. (Perhaps you took an OpenOffice table and used the table-to-text menu command to render the table as tab-delimited text.) Presumably there's a tab separating each columnar data item and a hard return at the end of each line. Perfect. You're ready to program.
Use Control-A to Select All of the table. (Or; Highlight the whole thing by swiping across the page with the mouse.) In the JS console, type
myTable = getSelection( ).toString( );
Now you have the whole table in one variable.
Alternatively, instead of selecting all, you could grab the entire text file programmatically this way:
wholePage = document.getElementsByTagName("pre")[0].innerHTML;
When you open a text file in a browser, it always puts the text in a <pre> element. The method getElementsByTagName( ) always returns an array, even if there's just one item in it (as in this case). To get the one and only item in the array, use index zero: [0]. To get the text of the element, use innerHTML.
To get the rows from your table:
rows = myTable.split("\n");
By splitting on every newline (carriage return), you get an array of table tows. To get the columns for row 7 (which is actually at index 6 in the array):
columns = rows[6].split("\t");
By splitting on tabs, you've created an array of column items. When you want to display something in the console, do
console.log( columns[ 0 ] );
In this case we've chosen to display the first column item (which is at index zero).
This is all familiar enough stuff to experienced JS users. I'm showing the basics here for those who might be new to programming. If you're new to programming, you need to be reminded (arguably) that array items are indexed in zero-based fashion: the first item is at index zero, the second item is at index one, the final item is always at length-minus-one.
If you're new to the Chrome JavaScript console (Control-Shift-J opens the console; plus be sure the Console tab is selected), you'll want to take note that you need to use Shift-Enter to type new lines in the console. Just hitting Enter executes whatever code is in the console. Write a line of code, then Shift-Enter to continue writing on a new line.
To recap: A quick way to crunch data is to put your data in a text file, open the text file with your browser, select all content, then use getSelection().toString() to load the selected content into a variable. Alternatively, load the whole text page with document.getElementsByTagName("pre")[0].innerHTML. From there, you can parse and crunch at will.
Wednesday, July 31, 2013
Tuesday, July 30, 2013
jQuery for Bioinformatics
I've been using JavaScript for almost two decades now, but somehow I've managed to avoid learning jQuery until just recently, mostly out of laziness but also because of a lingering yet torrid love-hate relationship with "syntax sugar" programming patterns. The best thing I can say about jQuery is that it has a seductively compact and powerful syntax. The worst thing I can say about jQuery is this.repeat(previousStatement).
For better or worse, I've had to begin dabbling in jQuery recently to save myself from the horror of old-school bare-knuckle DOM parsing. You know what I'm talking about: Nested loops with lots of calls to getElementsByTagName( ) followed up with hand-parsing of innerHTML. Who wants to do all that when you can use the oh-so-cute $(selector).each( ) construction?
The trouble with cute/compact syntax (as any recovering Perl user will gladly tell you in return for a bottle of cheap sherry) is that it's write-only. When you go back to look at something a week later and see 15 lines' worth of JS functionality rolled up into a shockingly crisp (yet thoroughly opaque) jQuery one-liner, you often wish you'd gone ahead and written those 15 homely lines of JavaScript in the first place, instead of giving in to that one irresistibly sexy, powerful line of jQuery that's oh yeah BTW also self-obfuscating.
Nonetheless, if you do a lot of page-scraping (as I do when visiting bioinformatics sites), the time savings of being able to parse a page with jQuery can be formidable. Who can resist grabbing all rows of a table with $("tr")? Who can resist iterating over them with .each()?
I tend to use the online apps at genomevolution.org quite heavily. The great folks who maintain that site have a nice way of serving up prodigious amounts of data in easy-to-use interactive forms, but sometimes you just want to harvest the data from a table and be done with it. Take the page I created at http://genomevolution.org/r/9726, which is based on a list of 100 unique bacterial species in the group known as Alphaproteobacteria. If you go to that page and scroll over to the far right, you'll see a column header labeled "Codon Usage." Underneath that label is a "Get All Codon Tables" link. Click that link and be prepared to wait about two minutes as the codon data loads for each organism. It's worth the wait, because when you're done, you're looking at color-coded codon usage frequencies for all 64 codons, for all 100 organisms.
Suppose you just want the codon data in text form, to analyze later? Scraping the raw data out of the HTML page is a royal bitch, because whether you know it or not, that page has tables embedded in tables embedded in tables. Parsing the DOM by hand is (shudder, wince) well nigh unthinkable.
Go to http://genomevolution.org/r/9726 and click "Get All Codon Tables" under the "Codon Usage" column heading. Allow a minute or two for codon data to load. Meanwhile, Control-Shift-J opens the Chrome console. (Select the Console tab at the top of the window if it's not already selected.) Paste the following code into the console. Hit Enter. Savor the power.
All of this was originally a single statement, with an inline callback function (in typical jQuery fashion). I decided to unroll it into more verbose, easier to understand form, lest my head explode two weeks from now trying to re-read and re-figure-out the code.
This bit of code does some pretty typical jQuery things, such as grab all rows of a table with $('tr'), except that in this case I most certainly do not want all rows of all tables in the HTML page (which would be hundreds of rows of extraneous stuff). The rows I need happen to have an "id" attribute with a value that begins with "gl." The construction $('tr[id^=gl]') is jQuery's syntax for selecting table rows that have an id-attribute that begins with "gl." (The ^= here means "begins with." You could signify "ends with" using $= instead of ^=.)
The process() callback fetches all table columns for the current row using the jQuery( 'td', this ) construction, which means gives me a jQuery object representing all "td" elements under the DOM node represented by this. In the callback context, this refers to the current jQuery node, not the window object or Function object. If you choose (as I did not) to declare your callback with arguments, as in function myCallback( argA, argB), then argA will be the index of the current item and argB will be this.
If you're wondering about the regex /(?=CCA)/, I need this because ordinarily the codon data would look like this:
Codon Usage: The Bacterial and Plant Plastid Code (transl_table=11) CCA(P) 1.18%CCG(P) 1.58%CCT(P) 1.17%CCC(P) 1.37%CGA(R) 0.32%CGG(R) 1.32%CGT(R) 1.82%CGC(R) 2.54%CAA(Q) 1.07%CAG(Q) 2.84%CAT(H) 1.59%CAC(H) 0.89%CTA(L) 0.48%CTG(L) 4.58%CTT(L) 1.96%CTC(L) 0.84%GCA(A) 2.94%GCG(A) 2.14%GCT(A) 2.31%GCC(A) 3.90%GGA(G) 0.90%GGG(G) 1.74%GGT(G) 2.11%GGC(G) 3.23%GAA(E) 3.92%GAG(E) 1.36%GAT(D) 3.76%GAC(D) 1.49%GTA(V) 1.08%GTG(V) 3.01%GTT(V) 2.19%GTC(V) 0.81%ACA(T) 1.82%ACG(T) 1.49%ACT(T) 0.57%ACC(T) 1.83%AGA(R) 0.30%AGG(R) 0.31%AGT(S) 0.61%AGC(S) 1.33%AAA(K) 2.01%AAG(K) 1.60%AAT(N) 1.39%AAC(N) 1.64%ATA(I) 0.59%ATG(M) 2.56%ATT(I) 2.88%ATC(I) 1.59%TCA(S) 0.65%TCG(S) 0.47%TCT(S) 1.37%TCC(S) 1.34%TGA(*) 0.14%TGG(W) 1.47%TGT(C) 0.46%TGC(C) 0.70%TAA(*) 0.14%TAG(*) 0.03%TAT(Y) 1.47%TAC(Y) 0.90%TTA(L) 0.61%TTG(L) 1.67%TTT(F) 2.41%TTC(F) 1.22%
Notice that first line ("Codon usage: The Bacterial [blah blah]"). I just want the codon data, not the leader line. But how to split off the codon data? Answer: Use a lookahead regular expression that doesn't consume the match. If you split on /CCA/ (the first codon) you will of course consume the CCA, never to be seen again. Instead, use (?=CCA), with parentheses (absolutely essential!) and the parser will look ahead to find an upcoming CCA, then stop and match the spot right before the CCA without consuming the CCA.
I'm sure a true jQuery expert can rewrite the foregoing code in a much more elegant, compact manner. For me, elegant and compact aren't always optimal. I've learned to value readable and self-documenting over elegant and opaque. Cute/sexy isn't always best. I'll take homely and straightforward any day.
For better or worse, I've had to begin dabbling in jQuery recently to save myself from the horror of old-school bare-knuckle DOM parsing. You know what I'm talking about: Nested loops with lots of calls to getElementsByTagName( ) followed up with hand-parsing of innerHTML. Who wants to do all that when you can use the oh-so-cute $(selector).each( ) construction?
The trouble with cute/compact syntax (as any recovering Perl user will gladly tell you in return for a bottle of cheap sherry) is that it's write-only. When you go back to look at something a week later and see 15 lines' worth of JS functionality rolled up into a shockingly crisp (yet thoroughly opaque) jQuery one-liner, you often wish you'd gone ahead and written those 15 homely lines of JavaScript in the first place, instead of giving in to that one irresistibly sexy, powerful line of jQuery that's oh yeah BTW also self-obfuscating.
Nonetheless, if you do a lot of page-scraping (as I do when visiting bioinformatics sites), the time savings of being able to parse a page with jQuery can be formidable. Who can resist grabbing all rows of a table with $("tr")? Who can resist iterating over them with .each()?
I tend to use the online apps at genomevolution.org quite heavily. The great folks who maintain that site have a nice way of serving up prodigious amounts of data in easy-to-use interactive forms, but sometimes you just want to harvest the data from a table and be done with it. Take the page I created at http://genomevolution.org/r/9726, which is based on a list of 100 unique bacterial species in the group known as Alphaproteobacteria. If you go to that page and scroll over to the far right, you'll see a column header labeled "Codon Usage." Underneath that label is a "Get All Codon Tables" link. Click that link and be prepared to wait about two minutes as the codon data loads for each organism. It's worth the wait, because when you're done, you're looking at color-coded codon usage frequencies for all 64 codons, for all 100 organisms.
Suppose you just want the codon data in text form, to analyze later? Scraping the raw data out of the HTML page is a royal bitch, because whether you know it or not, that page has tables embedded in tables embedded in tables. Parsing the DOM by hand is (shudder, wince) well nigh unthinkable.
Go to http://genomevolution.org/r/9726 and click "Get All Codon Tables" under the "Codon Usage" column heading. Allow a minute or two for codon data to load. Meanwhile, Control-Shift-J opens the Chrome console. (Select the Console tab at the top of the window if it's not already selected.) Paste the following code into the console. Hit Enter. Savor the power.
codonData = [];
function process( ) {
var CODONS_COLUMN = 15;
var rowdata = jQuery( 'td', this );
var codonUsage = rowdata[ CODONS_COLUMN ].textContent.split(/(?=CCA)/)[1];
codonData.push( codonUsage );
}
$('tr[id^=gl]').each( process ); // oh jQuery, must you tease me so?
console.log( codonData.join("\n") );
All of this was originally a single statement, with an inline callback function (in typical jQuery fashion). I decided to unroll it into more verbose, easier to understand form, lest my head explode two weeks from now trying to re-read and re-figure-out the code.
This bit of code does some pretty typical jQuery things, such as grab all rows of a table with $('tr'), except that in this case I most certainly do not want all rows of all tables in the HTML page (which would be hundreds of rows of extraneous stuff). The rows I need happen to have an "id" attribute with a value that begins with "gl." The construction $('tr[id^=gl]') is jQuery's syntax for selecting table rows that have an id-attribute that begins with "gl." (The ^= here means "begins with." You could signify "ends with" using $= instead of ^=.)
The process() callback fetches all table columns for the current row using the jQuery( 'td', this ) construction, which means gives me a jQuery object representing all "td" elements under the DOM node represented by this. In the callback context, this refers to the current jQuery node, not the window object or Function object. If you choose (as I did not) to declare your callback with arguments, as in function myCallback( argA, argB), then argA will be the index of the current item and argB will be this.
If you're wondering about the regex /(?=CCA)/, I need this because ordinarily the codon data would look like this:
Codon Usage: The Bacterial and Plant Plastid Code (transl_table=11) CCA(P) 1.18%CCG(P) 1.58%CCT(P) 1.17%CCC(P) 1.37%CGA(R) 0.32%CGG(R) 1.32%CGT(R) 1.82%CGC(R) 2.54%CAA(Q) 1.07%CAG(Q) 2.84%CAT(H) 1.59%CAC(H) 0.89%CTA(L) 0.48%CTG(L) 4.58%CTT(L) 1.96%CTC(L) 0.84%GCA(A) 2.94%GCG(A) 2.14%GCT(A) 2.31%GCC(A) 3.90%GGA(G) 0.90%GGG(G) 1.74%GGT(G) 2.11%GGC(G) 3.23%GAA(E) 3.92%GAG(E) 1.36%GAT(D) 3.76%GAC(D) 1.49%GTA(V) 1.08%GTG(V) 3.01%GTT(V) 2.19%GTC(V) 0.81%ACA(T) 1.82%ACG(T) 1.49%ACT(T) 0.57%ACC(T) 1.83%AGA(R) 0.30%AGG(R) 0.31%AGT(S) 0.61%AGC(S) 1.33%AAA(K) 2.01%AAG(K) 1.60%AAT(N) 1.39%AAC(N) 1.64%ATA(I) 0.59%ATG(M) 2.56%ATT(I) 2.88%ATC(I) 1.59%TCA(S) 0.65%TCG(S) 0.47%TCT(S) 1.37%TCC(S) 1.34%TGA(*) 0.14%TGG(W) 1.47%TGT(C) 0.46%TGC(C) 0.70%TAA(*) 0.14%TAG(*) 0.03%TAT(Y) 1.47%TAC(Y) 0.90%TTA(L) 0.61%TTG(L) 1.67%TTT(F) 2.41%TTC(F) 1.22%
Notice that first line ("Codon usage: The Bacterial [blah blah]"). I just want the codon data, not the leader line. But how to split off the codon data? Answer: Use a lookahead regular expression that doesn't consume the match. If you split on /CCA/ (the first codon) you will of course consume the CCA, never to be seen again. Instead, use (?=CCA), with parentheses (absolutely essential!) and the parser will look ahead to find an upcoming CCA, then stop and match the spot right before the CCA without consuming the CCA.
I'm sure a true jQuery expert can rewrite the foregoing code in a much more elegant, compact manner. For me, elegant and compact aren't always optimal. I've learned to value readable and self-documenting over elegant and opaque. Cute/sexy isn't always best. I'll take homely and straightforward any day.
Friday, July 26, 2013
Getting Started in Desktop Bioinformatics
I've spent about four months now exploring do-it-yourself desktop bioinformatics. Overall, I'm excited by what I've been able to do and I'm optimistic about the prospects for other do-it-yourself desktop-science geeks, because there are tons of great online tools for doing bio-sci and lots of important scientific questions yet to be fleshed out. So I thought I'd share some of what I've learned, and provide some pointers for anyone who might want to try his or her hand at this sort of "citizen science."
It helps to have the benefit of a science education (in particular, a bio education) before beginning, but one of the great things about do-it-yourself desktop science is that you can (and will!) learn as you go. For example, you might have only a bare-bones basic understanding of enzymology before you begin, but as you move deeper into a particular research quest, you'll find yourself wanting to learn more about this or that aspect of an enzyme. So you'll hit Google Scholar and bring yourself up-to-date on this or that detail of a particular subject. That's a Good Thing.
When I first plunged into DNA analysis, I have to admit my knowledge of mitochondria was weak. I knew they had their own DNA, for example, but it wasn't obvious to me (until I started digging) that mitochondrial DNA is pathetically small, whereas the mitochondrial proteome (the superset of all products that go into making up a functioning mitochondrion) is large. In other words, most "mitochondrial genes" are not in mtDNA. They're in nuclear DNA. There are a couple of online databases of nuclear mitochondrial genes (NUMTs, as they're known), but by and large this is an area in dire need of more research. Someone needs to put together a database or reference set of yeast NUMTs, for example. We also need a database for algal NUMTs, another for protozoan NUMTs, another for rice or corn or Arabidopsis NUMTs, etc. Maybe you'll be the one to move such a project along?
So. How can you get started in desktop bioinformatics? I recommend familiarizing yourself with the great tools at genomevolution.org, which is powered by iPlant, which (in turn) is funded by the National Science Foundation Plant Cyberinfrastructure Program here in the U.S. In particular, I recommend you set aside an evening to run through some of the tutorials at genomevolution.org. That'll give you an idea of what's possible with their tools.
If you go to this page and scroll down, you'll find some really interesting short videos showing how to use some of the genomevolution.org tools. They're fun to watch and should stimulate your imagination.
What kinds of problems need investigating by desktop biologists? The sky's the limit. One quest that lends itself to citizen science is looking for examples of horizontal gene transfer (HGT). This requires that you first teach yourself a little bit about BLAST searches. (BLAST searches are sequence-similarity searches that let you compare DNA against DNA or amino-acid sequence against amino-acid sequence.) The strategies involved here can range from simple and brute force to sophisticated; and the great thing is, you can invent your own heuristics. It's a wide-open area. I recently found good evidence (90%+ similarity of DNA sequences) for bacterial gene transfer into rice, which I'll write about in a later post. I'm confident there are thousands of examples of horizontal gene transfer (whether from bacteria to bacteria, bacteria to plant, bacteria to insect, or whatever) waiting to be discovered. You could easily be the next discoverer of one of these gene transfers.
Here are some other ideas for desktop-science explorations:
It helps to have the benefit of a science education (in particular, a bio education) before beginning, but one of the great things about do-it-yourself desktop science is that you can (and will!) learn as you go. For example, you might have only a bare-bones basic understanding of enzymology before you begin, but as you move deeper into a particular research quest, you'll find yourself wanting to learn more about this or that aspect of an enzyme. So you'll hit Google Scholar and bring yourself up-to-date on this or that detail of a particular subject. That's a Good Thing.
When I first plunged into DNA analysis, I have to admit my knowledge of mitochondria was weak. I knew they had their own DNA, for example, but it wasn't obvious to me (until I started digging) that mitochondrial DNA is pathetically small, whereas the mitochondrial proteome (the superset of all products that go into making up a functioning mitochondrion) is large. In other words, most "mitochondrial genes" are not in mtDNA. They're in nuclear DNA. There are a couple of online databases of nuclear mitochondrial genes (NUMTs, as they're known), but by and large this is an area in dire need of more research. Someone needs to put together a database or reference set of yeast NUMTs, for example. We also need a database for algal NUMTs, another for protozoan NUMTs, another for rice or corn or Arabidopsis NUMTs, etc. Maybe you'll be the one to move such a project along?
So. How can you get started in desktop bioinformatics? I recommend familiarizing yourself with the great tools at genomevolution.org, which is powered by iPlant, which (in turn) is funded by the National Science Foundation Plant Cyberinfrastructure Program here in the U.S. In particular, I recommend you set aside an evening to run through some of the tutorials at genomevolution.org. That'll give you an idea of what's possible with their tools.
 |
| Many organisms have genes for flagellum proteins, but not all such organisms actually make a flagellum. (The flagellum is the whiplike appendage that gives the cell motility. Above: Bdellovibrio, a bacterium with a powerful flagellum.) |
If you go to this page and scroll down, you'll find some really interesting short videos showing how to use some of the genomevolution.org tools. They're fun to watch and should stimulate your imagination.
What kinds of problems need investigating by desktop biologists? The sky's the limit. One quest that lends itself to citizen science is looking for examples of horizontal gene transfer (HGT). This requires that you first teach yourself a little bit about BLAST searches. (BLAST searches are sequence-similarity searches that let you compare DNA against DNA or amino-acid sequence against amino-acid sequence.) The strategies involved here can range from simple and brute force to sophisticated; and the great thing is, you can invent your own heuristics. It's a wide-open area. I recently found good evidence (90%+ similarity of DNA sequences) for bacterial gene transfer into rice, which I'll write about in a later post. I'm confident there are thousands of examples of horizontal gene transfer (whether from bacteria to bacteria, bacteria to plant, bacteria to insect, or whatever) waiting to be discovered. You could easily be the next discoverer of one of these gene transfers.
Here are some other ideas for desktop-science explorations:
- Find and characterize flagellar genes in organisms that lack motility. If you dig into the literature, you'll find that there are many examples of supposedly immotile organisms (like the intracellular parasite Buchnera, which lives inside aphids) that not only harbor flagellum genes but express some of them—yet have no external flagellum. Obviously, organisms that retain flagellum genes but actually don't make a flagellum (that little whip-like tail that makes single-celled organisms swim around) must be retaining those genes for a reason. The gene products must be doing something. But what? Also: Paramecium and diatoms and other eukaryotes make flagella and/or cilia. Most animals also make cilia. (Ever get a tickle deep in your throat or bronchia? It was probably something tangling with the cilia lining your bronchial system.) What's the relationship between cilia gene products in Paramecium, say, and cilia in animals? Do any plants conceal cilia genes? If so, how are they related phylogenetically to lower-organism cilia?
- Migration of genes from parasites to host DNA. A general pattern that seems to happen in nature is: a bacterium or other invader takes up residency inside an animal or plant cell, becoming an endosymbiont; then some of its genes (the symbiont's genes) move to the nucleus of the host cell. Which genes? What do the genes do? That's up to you to try to find out.
- Bidirectionally ("bidi") transcribed genes: While rare, there are examples of genes in which each strand of DNA is transcribed into mRNA. (The genome for Rothia mucilaginosa contains many putative examples of this.) Find organisms that contain bidi genes. Try to determine if both strands are actually transcribed. Examine sister-species organisms to see if one strand is transcribed in one organism and the other gene (on the other strand) is transcribed in the other organism.
- Phylogenetics of plasmid and viral genes. Try to determine the ancestry of a virus gene. There are good tree-making services online that do all the hard work for you, including protein-sequence alignments. All you have to do is cut and paste Fasta files.
- Codon analysis. There are many plants (rice is one) and higher organisms in which DNA is more or less equally divided into high-GC-content genes and low-GC-content genes. Surely the codon usage patterns for each class of gene(s) varies. But how? What are the codon adaptation indexes (CAI values) for the various genes? Create a few histograms of CAI values. Use CAIs and other techniques to try to determine which genes are highly expressed. Are HEGs (highly expressed genes) mostly high-GC? Low-GC? Both? Run some histograms on the genes' purine (A+G) content, G+C content, G+C content by codon position.
- Many organisms (and organelles) have extremely GC-poor genomes. Some have bizarre codon usage patterns (where, say, the codon AAA is used 12 times more than the average codon). Some use 56 or fewer codons (out of 64 possible). Find the organelle or organism that uses the fewest codons. See if there's an organism or organelle that uses fewer than 20 amino acids. Which amino acid(s) get(s) left out?
- Characterize the DNA repairosome of an aerobic and an anerobic archeon. Compare and contrast the two.
- Find all the genes in a particular organism that have mitochondrial-targeting presequences in their DNA.
- Pick two closely related organisms. Try to figure out how many million years ago they diverged. Use mitochondrial DNA analysis as well as cytoplasmic protein analysis.
- Find the bacterium that has more secretion-protein, permease, and protein-translocation genes than any other. Compare it to its closest relative.
- Find an organism that is pathogenic (to humans, animals, or plants). Find its closest non-pathogenic relative. Compare genomes. Determine which genes are most likely to be involved in virulence.
- Some seemingly simple organisms (amoebae) have more DNA than a human. Why? What's all that DNA doing there? Does it contain horizontally transferred genes from plants, bacteria, archeons, animals? Are amoebas and other super-large-genome organisms "DNA hoarders"? Are they DNA curationists? Characterize the genes (enumerate by category, first) of these organisms. How many are expressed? How many are junk? What's the energy cost of maintaining that much junk DNA? Can it all be junk? Is an amoeba actually a devolved higher life form that forgot how to do morphogenesis and can no longer develop into a tadpole or whatever?
- [ insert your own project here! ]
Sunday, July 21, 2013
Why Mitochondrial DNA is Different
Most genomes that are high in A+T content (or low in G+C content) show a surprising DNA strand asymmetry: The message strand of genes tends to be rich in purines. This rule applies across all domains I've looked at except mitochondria, where message strands tend to be pyrimidine-rich rather than purine-rich. The following two graphs makes this clearer.

This is a graph of message-strand (or RNA-synonymous-strand) purine content plotted vertically, against A+T plotted horizontally, for 1,373 bacterial species. Each dot represents a genome. High-GC/low-AT organisms like Streptomyces and Bordetella are on left and low-GC/high-AT organisms like Clostridium botulinum are toward the right. The few dots on the far right are intracellular endosymbionts that have lost a good bit of DNA over the millennia. They tend to be extremely high in A+T.
Compare the above graph with the graph below, which is the same thing (message-strand A+G vs. A+T) for mitochondrial DNA (N=2543 genomes). There is still an upward slope to the data (and in fact it is steeper than it looks, because the range of y-values is different in the graph below than in the graph above). The slope of the regression line is very nearly the same (0.148 vs. 0.149) for both graphs. But you can see that in the graph below, nearly all the points are below y = 0.50. That means message-strands are high in pyrimidines rather than purines.

I speculated in a previous post that the reason mitochondrial DNA is pyrimidine-heavy on the message strand is that mtDNA encodes a very small number of proteins (13, in all), and they tend to be membrane-associated proteins, which use mostly non-polar amino acids. It turns out that codons for the non-polar amino acids are pyrimidine-rich.
To see if that's really what's going on, I obtained the DNA sequences for cytochrome-c oxidase and NADH dehydrogenase (the two must fundamental enzyme systems of mitochondria) from several hundred bacterial species. Actually, I was able to obtain DNA sequences for a total of 942 bacterial NADH dehydrogenase (subunit L) proteins. I also succeeded in obtaining DNA sequences for 647 bacterial cytochrome-c oxidase subunit 1 proteins. In mitochondria, these genes are known as ND5 and Cox1. In bacteria they're better known as nuoL and cyoB.
The graph below shows A+G for the two enzymes versus whole-chromosome A+T, for the relevant organisms.
The pink points are for cytochrome-c oxidase subunit 1 (cyoB) while the blue points are for NADH dehydrogenase subunit 5 (nuoL). Two things are worth noting. One is that the regression line is upward-sloping, meaning that as an organism's DNA gets richer in A+T content, the purine content on the message strand rises. This effect seems to be universal. The second thing to note is that almost all of the points in the graph lie below y = 0.5, as is the case for mitochondria. These two signature "mitochondrial" enzyme systems, critical to oxidative phosphorylation (in bacteria as well as higher organisms), do tend to use pyrimidine-rich codons—rendering the relevant genes pyrminidine-rich on the RNA-synonymous (message) strand of DNA. The hypothesis is upheld.
For you bio students, a bit of homework: You might want to think about why it is that membrane-associated proteins are rich in non-polar amino acids. (In human mitochondria, leucine and isoleucine are the most-used amino acids. Together they account for an amazing 30% of all amino acids used in mtDNA-encoded gene products.) Hint: Most membranes have a lipid bilayer, and lipids don't like water.

This is a graph of message-strand (or RNA-synonymous-strand) purine content plotted vertically, against A+T plotted horizontally, for 1,373 bacterial species. Each dot represents a genome. High-GC/low-AT organisms like Streptomyces and Bordetella are on left and low-GC/high-AT organisms like Clostridium botulinum are toward the right. The few dots on the far right are intracellular endosymbionts that have lost a good bit of DNA over the millennia. They tend to be extremely high in A+T.
Compare the above graph with the graph below, which is the same thing (message-strand A+G vs. A+T) for mitochondrial DNA (N=2543 genomes). There is still an upward slope to the data (and in fact it is steeper than it looks, because the range of y-values is different in the graph below than in the graph above). The slope of the regression line is very nearly the same (0.148 vs. 0.149) for both graphs. But you can see that in the graph below, nearly all the points are below y = 0.50. That means message-strands are high in pyrimidines rather than purines.

I speculated in a previous post that the reason mitochondrial DNA is pyrimidine-heavy on the message strand is that mtDNA encodes a very small number of proteins (13, in all), and they tend to be membrane-associated proteins, which use mostly non-polar amino acids. It turns out that codons for the non-polar amino acids are pyrimidine-rich.
To see if that's really what's going on, I obtained the DNA sequences for cytochrome-c oxidase and NADH dehydrogenase (the two must fundamental enzyme systems of mitochondria) from several hundred bacterial species. Actually, I was able to obtain DNA sequences for a total of 942 bacterial NADH dehydrogenase (subunit L) proteins. I also succeeded in obtaining DNA sequences for 647 bacterial cytochrome-c oxidase subunit 1 proteins. In mitochondria, these genes are known as ND5 and Cox1. In bacteria they're better known as nuoL and cyoB.
The graph below shows A+G for the two enzymes versus whole-chromosome A+T, for the relevant organisms.
 |
| Message strand purine content was derived from the DNA sequences of cyoB (pink) genes from 942 bacteria, and from nuoL (blue) genes from 647 bacterial species. The A+G values were plotted against host-organism whole-genome A+T content. All cyoB and nuoL sequences tended to be pyrimidine rich. But pyrimidine content was less for organisms with high A+T content. (Note the slightly positive slope of the regression line.) |
The pink points are for cytochrome-c oxidase subunit 1 (cyoB) while the blue points are for NADH dehydrogenase subunit 5 (nuoL). Two things are worth noting. One is that the regression line is upward-sloping, meaning that as an organism's DNA gets richer in A+T content, the purine content on the message strand rises. This effect seems to be universal. The second thing to note is that almost all of the points in the graph lie below y = 0.5, as is the case for mitochondria. These two signature "mitochondrial" enzyme systems, critical to oxidative phosphorylation (in bacteria as well as higher organisms), do tend to use pyrimidine-rich codons—rendering the relevant genes pyrminidine-rich on the RNA-synonymous (message) strand of DNA. The hypothesis is upheld.
For you bio students, a bit of homework: You might want to think about why it is that membrane-associated proteins are rich in non-polar amino acids. (In human mitochondria, leucine and isoleucine are the most-used amino acids. Together they account for an amazing 30% of all amino acids used in mtDNA-encoded gene products.) Hint: Most membranes have a lipid bilayer, and lipids don't like water.
Saturday, July 20, 2013
More about Mitochondrial DNA
To recap my desktop-science experiments of the last month or so, I've found strandwise DNA asymmetry across domains, which is to say in bacteria, Archaea, eukaryotes, viruses, and mitochondrial DNA. In every case except mitochondria, the message (or RNA-synonymous) strand of DNA in coding regions tends to be purine-rich. The opposite strand tends to be pyrimidine-rich. Moreover, in all domains, including mitochondria, message-strand purine content increases in proportion to genome A+T content. (A+T content is a phylogenetic signature. Some genomes are inherently high in A+T content—or low in G+C content—while others are not. Related organisms tend to have similar A+T or G+C contents.)
Mitochondrial genes tend to be pyrmidine-rich on the message strand, seemingly in violation of the finding that in all other domains, message strands are purine-rich. The mitochondrial anomaly is actually very easy to understand (although it took me weeks to realize the explanation). In a nutshell: Mitochondrial DNA is pyrimidine-rich on message strands because mtDNA encodes only a few proteins (13, usually), all of them membrane-associated. Membrane-associated proteins are unusual because they tend to incorporate mostly non-polar amino acids such as leucine, isoleucine, valine, proline, alanine, or phenylalanine—all of which are specified by pyrimidine-rich codons.
It seems to me mitochondrial DNA shouldn't be thought of as a genome, because well over 90% of mitochondrial-associated gene products are encoded by genes in the host nucleus. (In humans, there may be as many as 1500 nuclear-encoded mitochondrial genes.) This point is worth repeating, so let me quote Patrick Chinnery, TRENDS in Genetics (2003) 19:2, 60:
The circular mitochondrial "chromosome" (if it can be called that) is the vestigial remnant of a much larger genome that long ago migrated to the host nucleus, no doubt to avoid oxidative attack. The mitochondrion simply is not a safe place to store DNA. (Would you set up a sperm bank in a rocket-fuel factory?) It's teeming with molecular oxygen, superoxides, peroxides, free protons, and other hazardous materials.
Human mitochondrial DNA (which is typical of a lot of mtDNA) encodes just a handful of multi-subnit transmembrane proteins, namely: cytochrome-c oxidase, NADH dehydrogenase, cytochrome-b, and an ATPase. That's it. There are no other protein genes in human mtDNA. All other "mitochondrial proteins" are encoded somewhere else. (That includes 37 out of 44 subunits of the NADH dehydrogenase complex; the DNA polymerase that replicates mitochondrial DNA; the mitochondrial RNA polymerase; about 50 ribosomal proteins; so-called "mitochondrial" catalase; and hundreds of other "mitochondrial" proteins. All are encoded in the nucleus.)
Bottom line: Mitochondrial DNA encodes a very small ensemble of highly specialized membrane-associated proteins. We shouldn't expect this small ensemble to be representative of other genes found in other genomes. (And it's not.) That, in a nutshell, is why mtDNA is not particularly purine-rich in message strands.
But we should test this hypothesis, if possible. (And it is, in fact, possible.) Most bacteria are aerobic, which means most bacterial species have genes for cytochrome-c oxidase, NADH dehydrogenase, etc. The DNA for those genes should be similar to mtDNA with respect to strand-asymmetric purine content. If we analyze bacterial DNA, we should find that genes for cytochrome-c oxidase, NADH dehydrogenase, etc. are pyrimidine-rich on the message strand, just as in mtDNA.
In tomorrow's post: the data.
Mitochondrial genes tend to be pyrmidine-rich on the message strand, seemingly in violation of the finding that in all other domains, message strands are purine-rich. The mitochondrial anomaly is actually very easy to understand (although it took me weeks to realize the explanation). In a nutshell: Mitochondrial DNA is pyrimidine-rich on message strands because mtDNA encodes only a few proteins (13, usually), all of them membrane-associated. Membrane-associated proteins are unusual because they tend to incorporate mostly non-polar amino acids such as leucine, isoleucine, valine, proline, alanine, or phenylalanine—all of which are specified by pyrimidine-rich codons.
 |
| The mitochondrion. |
It seems to me mitochondrial DNA shouldn't be thought of as a genome, because well over 90% of mitochondrial-associated gene products are encoded by genes in the host nucleus. (In humans, there may be as many as 1500 nuclear-encoded mitochondrial genes.) This point is worth repeating, so let me quote Patrick Chinnery, TRENDS in Genetics (2003) 19:2, 60:
The vast majority of mitochondrial proteins (estimated at >1000) are synthesized in the cytosol from nuclear gene transcripts.
The circular mitochondrial "chromosome" (if it can be called that) is the vestigial remnant of a much larger genome that long ago migrated to the host nucleus, no doubt to avoid oxidative attack. The mitochondrion simply is not a safe place to store DNA. (Would you set up a sperm bank in a rocket-fuel factory?) It's teeming with molecular oxygen, superoxides, peroxides, free protons, and other hazardous materials.
 |
| The human mitochondrial chromosome. |
Human mitochondrial DNA (which is typical of a lot of mtDNA) encodes just a handful of multi-subnit transmembrane proteins, namely: cytochrome-c oxidase, NADH dehydrogenase, cytochrome-b, and an ATPase. That's it. There are no other protein genes in human mtDNA. All other "mitochondrial proteins" are encoded somewhere else. (That includes 37 out of 44 subunits of the NADH dehydrogenase complex; the DNA polymerase that replicates mitochondrial DNA; the mitochondrial RNA polymerase; about 50 ribosomal proteins; so-called "mitochondrial" catalase; and hundreds of other "mitochondrial" proteins. All are encoded in the nucleus.)
Bottom line: Mitochondrial DNA encodes a very small ensemble of highly specialized membrane-associated proteins. We shouldn't expect this small ensemble to be representative of other genes found in other genomes. (And it's not.) That, in a nutshell, is why mtDNA is not particularly purine-rich in message strands.
But we should test this hypothesis, if possible. (And it is, in fact, possible.) Most bacteria are aerobic, which means most bacterial species have genes for cytochrome-c oxidase, NADH dehydrogenase, etc. The DNA for those genes should be similar to mtDNA with respect to strand-asymmetric purine content. If we analyze bacterial DNA, we should find that genes for cytochrome-c oxidase, NADH dehydrogenase, etc. are pyrimidine-rich on the message strand, just as in mtDNA.
In tomorrow's post: the data.
Friday, July 19, 2013
Strand Asymmetry in Mitochondrial DNA
Funny how the availability of so much free DNA data can go to your head. When I learned that DNA sequence data for more than 2,000 mitochondrial genomes could be accessed, free, at genomevolution.org, I couldn't resist: I wrote some scripts that checked the DNA composition of 2,543 mtDNA (mitochondrial DNA) sequences. What I found blew me away.
If you're a biologist, you're accustomed to thinking of genome G+C (guanine plus cytosine) content as a kind of phylogenetic signature. (Related organisms usually have G+C values that are fairly close to one another.) For purposes of the following discussion, I'm going to reference A+T content, which is, of course, just one-minus-GC. (A GC content of 0.25, or 25%, means the AT content is 0.75, or 75%).
What I learned is that mitochondrial DNA shows strand asymmetry in coding regions (regions that actually get transcribed to RNA, as opposed to non-coding "control" regions and junk DNA). In particular, it shows an excess of pyrimidines (T and C) on the "message strand." This is the exact opposite of the situation in Archaea and bacteria, where message strands tend to accumulate purines (G and A).
The interesting thing is, just like bacteria (and Archaea), mitochondrial genomes tend to show a steady, predictable rate of increase of purines on the message strand with increasing A+T, even though purines are outnumbered by pyrimidines on the message strand. A picture might make this clearer:
Every point in this graph represents a mitochondrial genome (2,543 in all). As you can see, the regression line (which minimizes the sum of squared error) is upward-sloping, with a rise of 0.149, meaning that for every 10% increase in genome A+T content, there's a corresponding 1.49% increase in message-strand purine (A+G) content. What's striking about this is that in a similar graph for 1,373 bacterial genomes (see this post), the regression-line slope turned out to be 0.148. Chargaff's second parity law predicts a straight horizontal line at y=0.5. Obviously that law is kaput.
I've written before about my repeated finding (in bacteria, Archaea, eukaryotes, viruses, bacteriophage; basically every place I look) that message-strand purine content accumulates in proportion to genome A+T content. Strand asymmetry with respect to purines and pyrimidines seems to be universal. But why?
Strand-asymmetric buildup of purines or pyrimidines is very hard to explain without invoking either a theory of strand-asymmetric DNA repair or a theory of strand-asymmetric mutagenesis, or both. Is it reasonable to suppose that one strand of DNA is more vulnerable to mutagenesis than another? Yes, if you accept that in a growing cell, the strands spend a good portion of their time apart (during transcription and replication). Neither replication nor transcription is symmetric in implementation. I'll spare you the details for the replication side of the argument, but suffice it to say, replication-related asymmetries are not likely (in my opinion) to be behind the purine/pyrminidine strand asymmetries I've been documenting. What we're seeing, I think, is the result of asymmetric repair at transcription time.
During transcription, a gene's DNA strands are separated. One strand is used as a template by RNA polymerase to create messenger RNA and ribosomal RNA. The other strand is free and floppy and vulnerable to attack by mutagens. But it's also readily accessible to repair enzymes.

The above diagram oversimplifies things considerably, but I include it for the benefit of non-biogeeks who might want to follow this argument through. Note that DNA strands have directionality: the sugar bonds face one way in one strand and the other way in the other strand. This is denoted by the so-called 5'-to'3 orientation of strands.(RNAP = RNA polymerase.)
DNA repair is a complex subject. Be assured, every cell, of every kind, has dozens of different kinds of enzymes devoted to DNA repair. Without these enzymes, life as we know it would end, because DNA is constantly undergoing attack and requiring repair.
Some types of repair take place in double-stranded DNA (that is, DNA that is not undergoing replication or transcription). Other types of repair apply to single-stranded DNA. In bacteria as well as higher life forms, there's a transcription-coupled repair system (TCRS) that comes into play when RNA polymerase is stalled by thymine dimers or other DNA damage. This remarkably elaborate system changes out short sections of damaged DNA (at considerable energy cost). Because it involves replacing whole nucleotides (sugar and all), it's categorized as a Nucleotide Execision Repair system (NER). The alternative to NER is Base Excision Repair (BER), which is where a defective base (usually an oxidized guanine) gets snipped out without removing any sugars from the DNA backbone. The enzymes that perform this base-clipping are generically known as glycosylases.
For many years, it was thought that mitochondria did not have DNA repair systems. We now know that's not true. Mitochondrial DNA is subject to constant oxidative attack and it turns out the damage is quickly repaired, in double-stranded DNA. Evidence for repair of single-stranded mtDNA is scant. Those who have looked for a transcription-coupled repair system (or indeed any NER system) in mitochondria have not found one. Mitochondrial BER repair (via Ogg1) does exist, but it seems to operate when the DNA is double-stranded, not during transcription. This makes sense, because for BER to finish, the strand must be nicked by AP endonuclease after the bad base is popped out, then the repair proceeds by matching the opposing base (opposite the abasic site) using the other strand as template. In Clostridia and Archaea (which have an Ogg enzyme that other bacteria do not have; see this post and this paper), Ogg1 can pop out a bad base while the DNA is single-stranded; Ogg1 then binds to the abasic site and is only released by AP endonuclease when it arrives later on.
Bottom line, we know that mitochondrial DNA spends much of its time in the unwound state (because mtDNA products are very highly transcribed) and that the non-transcribed DNA strand is extremely vulnerable to oxidative attack. (The template strand is less vulnerable, because it is cloaked in enzymes: RNA polymerase, transcription factors, ribosomes, etc.) We also know that 8-oxoguanine is the most prevalent form of oxidative damage in mtDNA and that, uncorrected, such damage leads to G-to-T transversion. The finding of consistently high pyrimidine content in the message strand of mitochondrial DNA (see graph further above) is consistent with a slower rate of repair of the non-transcribed strand, and the differential occurrence of G-to-T transversions on that strand. Or at least, that's a possible explanation of the pyrimidine richness of the message strand of mtDNA.
But there are additional factors to consider, such as selection pressure. Mitochondrial DNA tends to encode membrane-associated proteins, and membrane proteins use nonpolar amino acids, which are (in turn) predominantly encoded by pyrimidine-rich codons. More about this in an upcoming post.
If you're a biologist, you're accustomed to thinking of genome G+C (guanine plus cytosine) content as a kind of phylogenetic signature. (Related organisms usually have G+C values that are fairly close to one another.) For purposes of the following discussion, I'm going to reference A+T content, which is, of course, just one-minus-GC. (A GC content of 0.25, or 25%, means the AT content is 0.75, or 75%).
What I learned is that mitochondrial DNA shows strand asymmetry in coding regions (regions that actually get transcribed to RNA, as opposed to non-coding "control" regions and junk DNA). In particular, it shows an excess of pyrimidines (T and C) on the "message strand." This is the exact opposite of the situation in Archaea and bacteria, where message strands tend to accumulate purines (G and A).
The interesting thing is, just like bacteria (and Archaea), mitochondrial genomes tend to show a steady, predictable rate of increase of purines on the message strand with increasing A+T, even though purines are outnumbered by pyrimidines on the message strand. A picture might make this clearer:
 |
| Purine (A+G) content versus A+T for the message strand of mitochondrial DNA coding regions (N=2543). |
Every point in this graph represents a mitochondrial genome (2,543 in all). As you can see, the regression line (which minimizes the sum of squared error) is upward-sloping, with a rise of 0.149, meaning that for every 10% increase in genome A+T content, there's a corresponding 1.49% increase in message-strand purine (A+G) content. What's striking about this is that in a similar graph for 1,373 bacterial genomes (see this post), the regression-line slope turned out to be 0.148. Chargaff's second parity law predicts a straight horizontal line at y=0.5. Obviously that law is kaput.
I've written before about my repeated finding (in bacteria, Archaea, eukaryotes, viruses, bacteriophage; basically every place I look) that message-strand purine content accumulates in proportion to genome A+T content. Strand asymmetry with respect to purines and pyrimidines seems to be universal. But why?
Strand-asymmetric buildup of purines or pyrimidines is very hard to explain without invoking either a theory of strand-asymmetric DNA repair or a theory of strand-asymmetric mutagenesis, or both. Is it reasonable to suppose that one strand of DNA is more vulnerable to mutagenesis than another? Yes, if you accept that in a growing cell, the strands spend a good portion of their time apart (during transcription and replication). Neither replication nor transcription is symmetric in implementation. I'll spare you the details for the replication side of the argument, but suffice it to say, replication-related asymmetries are not likely (in my opinion) to be behind the purine/pyrminidine strand asymmetries I've been documenting. What we're seeing, I think, is the result of asymmetric repair at transcription time.
During transcription, a gene's DNA strands are separated. One strand is used as a template by RNA polymerase to create messenger RNA and ribosomal RNA. The other strand is free and floppy and vulnerable to attack by mutagens. But it's also readily accessible to repair enzymes.

The above diagram oversimplifies things considerably, but I include it for the benefit of non-biogeeks who might want to follow this argument through. Note that DNA strands have directionality: the sugar bonds face one way in one strand and the other way in the other strand. This is denoted by the so-called 5'-to'3 orientation of strands.(RNAP = RNA polymerase.)
DNA repair is a complex subject. Be assured, every cell, of every kind, has dozens of different kinds of enzymes devoted to DNA repair. Without these enzymes, life as we know it would end, because DNA is constantly undergoing attack and requiring repair.
 |
| The Ogg family of DNA base-excision enzymes exhibit a signature helix-hairpin-helix topology (HhH). See Faucher et al., Int J Mol Sci 2012; 13(6): 6711–6729. |
For many years, it was thought that mitochondria did not have DNA repair systems. We now know that's not true. Mitochondrial DNA is subject to constant oxidative attack and it turns out the damage is quickly repaired, in double-stranded DNA. Evidence for repair of single-stranded mtDNA is scant. Those who have looked for a transcription-coupled repair system (or indeed any NER system) in mitochondria have not found one. Mitochondrial BER repair (via Ogg1) does exist, but it seems to operate when the DNA is double-stranded, not during transcription. This makes sense, because for BER to finish, the strand must be nicked by AP endonuclease after the bad base is popped out, then the repair proceeds by matching the opposing base (opposite the abasic site) using the other strand as template. In Clostridia and Archaea (which have an Ogg enzyme that other bacteria do not have; see this post and this paper), Ogg1 can pop out a bad base while the DNA is single-stranded; Ogg1 then binds to the abasic site and is only released by AP endonuclease when it arrives later on.
Bottom line, we know that mitochondrial DNA spends much of its time in the unwound state (because mtDNA products are very highly transcribed) and that the non-transcribed DNA strand is extremely vulnerable to oxidative attack. (The template strand is less vulnerable, because it is cloaked in enzymes: RNA polymerase, transcription factors, ribosomes, etc.) We also know that 8-oxoguanine is the most prevalent form of oxidative damage in mtDNA and that, uncorrected, such damage leads to G-to-T transversion. The finding of consistently high pyrimidine content in the message strand of mitochondrial DNA (see graph further above) is consistent with a slower rate of repair of the non-transcribed strand, and the differential occurrence of G-to-T transversions on that strand. Or at least, that's a possible explanation of the pyrimidine richness of the message strand of mtDNA.
But there are additional factors to consider, such as selection pressure. Mitochondrial DNA tends to encode membrane-associated proteins, and membrane proteins use nonpolar amino acids, which are (in turn) predominantly encoded by pyrimidine-rich codons. More about this in an upcoming post.
Wednesday, July 17, 2013
Highly Expressed Genes: Better-Repaired?
At any given time in any cell, some genes are highly expressed while others are moderately expressed, still others are barely expressed, and quite a few are not expressed at all. The fact that genes vary tremendously in their levels of expression is nothing new, of course, but we still have a lot to learn about how and why some genes have the "Transcribe Me!" knob cranked wide open and others remain dormant until called upon. (For a great paper on this subject, I recommend Samuel Karlin and Jan Mrázek, "Predicted Highly Expressed Genes of Diverse Prokaryotic Genomes," J. Bact. 2000 182:18, 5238-5250, free copy here.)
Reading up on this subject got me to thinking: If DNA undergoes damage and repair at transcription time (when genes are being expressed), shouldn't highly expressed genes differ in mutation rate from rarely expressed genes? (But, in which direction?) Also: Does one strand of highly expressed DNA (the strand that gets transcribed) mutate or repair at a different rate than the other strand?
We know that in most organisms, there is quite an elaborate repair apparatus dedicated to fixing DNA glitches at transcription time. (This is the so-called Transcription Coupled Repair System.) We also know that the TCRS has a preference for the template strand of DNA, just as RNA polymerase does. In fact, it's when RNA polymerase stalls at the site of a thymine dimer (or other major DNA defect) that TCRS kicks into action. Stalled RNAP is the trigger mechanism for TCRS.
But TCRS isn't the only repair option for DNA at transcription time. I've written before about the Archaeal Ogg1 enzyme (which detects and snips out oxidized guanine residues from DNA). The Ogg1 system is a much simpler Base Excision Repair system, fundamentally low-tech compared to the heavy-duty TCRS mechanism. The latter involves nucleotide-excision repair (NER), which means cutting sugars (deoxyribose) out of the DNA backbone and replacement of a whole section of DNA (at great energy cost). BER just snips bases and leaves the underlying sugar(s) in place.
Being a fan of desktop science, I wanted to see if I couldn't devise an experiment of my own to shed light on the question: Does differential repair of DNA strands at transcription time lead to strand asymmetry in highly expressed genes?
Happily, there's a database of highly expressed genes at http://genomes.urv.cat/HEG-DB, which is the perfect starting point for this sort of investigation. For my experiment, I chose the microbe Methanococcus maripaludis strain C5, This tiny organism (isolated from a salt marsh in South Carolina) is a strict anaerobe that lives off hydrogen gas and carbon dioxide. It has a relatively small genome (just under 1.7 million base pairs, enough to code for around 1400 genes). The complete genome is available from here (but don't click unless you want to start a 2-meg download). More to the point, a list of 123 of the creature's most highly expressed genes (HEGs) is available from this page (safe to click; no downloads). The HEGs are putative HEGs inferred from Codon Adaptation Index analysis relative to a reference set of (known-good) high-expression genes. For more details on the HEG ranking process see this excellent paper.
The DNA sequence data for M. maripaludis was easy to match up against the list of HEGs obtained from http://genomes.urv.cat/HEG-DB. In fact, I was able to do all the data-crunching I needed to do with a few lines of JavaScript, in the Chrome console. In no time, I had the adenine (A), guanine (G), and thymine (T) content for all of M. maripaludis's genes, which allowed me to make the following graph:
What we're looking at here is message-strand purine content (A+G) on the y-axis versus A+T content (which is a common phylogenetic metric, akin to G+C content) on the x-axis. As you know if you've been following this blog, I have used purine-vs.-AT plots quite successfully to uncover coding-region strand asymmetries. (See this post and/or this one for details.) The important thing to notice above is that while points tend to fall in a shotgun-blast centered roughly at x=0.66 and y=0.55, the Highly Expressed Genes (HEGs, in red) cover the upper left quadrant of the shotgun blast.
What does it mean? Consider the following. Of the four bases in DNA, guanine (G) is the most vulnerable to oxidative damage. When such damage is left uncorrected, it eventually results in a G-to-T transversion mutation. A large number of such mutations will cause overall A+T to increase (shifting points on the above graph to the right). If G-to-T transversions accumulate preferentially on one strand, the strand in question will see a reduction in purine content (as G, a purine, is replaced by T, a pyrimidine) while the other strand will see a corresponding increase in purine content (via the addition of adenines to pair with the new T's). Bottom line, if G-to-T transversions happen on the message strand, points in the above graph will move to the right and down. If they happen on the template (or transcribed) strand, points will move left and up. What we see in this graph is that HEGs have gone left and up.
The fact that highly expressed genes appear in the upper left quadrant of the distribution means that yes, differential repair is indeed (apparently) happening at transcription time; highly expressed genes are more intensively repaired; and the beneficiary of said repair(s), at least in M. maripaludis, is the message strand (also called the RNA-synonymous or non-transcribed strand) of DNA, which is where our sequence data come from, ultimately. A relative excess of unrepaired 8-oxoguanine on the template strand (or transcribed strand) means guanines are being replaced by thymines on that strand, and new adenines are showing up opposite the thymines, on the message strand, boosting A+G.
I don't know too many other explanations that are consistent with the above graph.
I hasten to add that one graph is just one graph. A single graph isn't enough to prove any kind of universal phenomenon. What we see here applies to Methanococcus maripaludis, an Archaeal anaerobe that may or may not share similarities (vis-a-vis DNA repair) with other organisms.
Reading up on this subject got me to thinking: If DNA undergoes damage and repair at transcription time (when genes are being expressed), shouldn't highly expressed genes differ in mutation rate from rarely expressed genes? (But, in which direction?) Also: Does one strand of highly expressed DNA (the strand that gets transcribed) mutate or repair at a different rate than the other strand?
We know that in most organisms, there is quite an elaborate repair apparatus dedicated to fixing DNA glitches at transcription time. (This is the so-called Transcription Coupled Repair System.) We also know that the TCRS has a preference for the template strand of DNA, just as RNA polymerase does. In fact, it's when RNA polymerase stalls at the site of a thymine dimer (or other major DNA defect) that TCRS kicks into action. Stalled RNAP is the trigger mechanism for TCRS.
But TCRS isn't the only repair option for DNA at transcription time. I've written before about the Archaeal Ogg1 enzyme (which detects and snips out oxidized guanine residues from DNA). The Ogg1 system is a much simpler Base Excision Repair system, fundamentally low-tech compared to the heavy-duty TCRS mechanism. The latter involves nucleotide-excision repair (NER), which means cutting sugars (deoxyribose) out of the DNA backbone and replacement of a whole section of DNA (at great energy cost). BER just snips bases and leaves the underlying sugar(s) in place.
Being a fan of desktop science, I wanted to see if I couldn't devise an experiment of my own to shed light on the question: Does differential repair of DNA strands at transcription time lead to strand asymmetry in highly expressed genes?
 |
| Methanococcus maripaludis |
The DNA sequence data for M. maripaludis was easy to match up against the list of HEGs obtained from http://genomes.urv.cat/HEG-DB. In fact, I was able to do all the data-crunching I needed to do with a few lines of JavaScript, in the Chrome console. In no time, I had the adenine (A), guanine (G), and thymine (T) content for all of M. maripaludis's genes, which allowed me to make the following graph:
 |
| Purine content (y-axis) plotted against adenine-plus-thymine content for all genes of Methanococcus maripaludis. Each dot represents a gene. The red dots represent the most highly expressed genes. Click to enlarge. |
What we're looking at here is message-strand purine content (A+G) on the y-axis versus A+T content (which is a common phylogenetic metric, akin to G+C content) on the x-axis. As you know if you've been following this blog, I have used purine-vs.-AT plots quite successfully to uncover coding-region strand asymmetries. (See this post and/or this one for details.) The important thing to notice above is that while points tend to fall in a shotgun-blast centered roughly at x=0.66 and y=0.55, the Highly Expressed Genes (HEGs, in red) cover the upper left quadrant of the shotgun blast.
What does it mean? Consider the following. Of the four bases in DNA, guanine (G) is the most vulnerable to oxidative damage. When such damage is left uncorrected, it eventually results in a G-to-T transversion mutation. A large number of such mutations will cause overall A+T to increase (shifting points on the above graph to the right). If G-to-T transversions accumulate preferentially on one strand, the strand in question will see a reduction in purine content (as G, a purine, is replaced by T, a pyrimidine) while the other strand will see a corresponding increase in purine content (via the addition of adenines to pair with the new T's). Bottom line, if G-to-T transversions happen on the message strand, points in the above graph will move to the right and down. If they happen on the template (or transcribed) strand, points will move left and up. What we see in this graph is that HEGs have gone left and up.
The fact that highly expressed genes appear in the upper left quadrant of the distribution means that yes, differential repair is indeed (apparently) happening at transcription time; highly expressed genes are more intensively repaired; and the beneficiary of said repair(s), at least in M. maripaludis, is the message strand (also called the RNA-synonymous or non-transcribed strand) of DNA, which is where our sequence data come from, ultimately. A relative excess of unrepaired 8-oxoguanine on the template strand (or transcribed strand) means guanines are being replaced by thymines on that strand, and new adenines are showing up opposite the thymines, on the message strand, boosting A+G.
I don't know too many other explanations that are consistent with the above graph.
I hasten to add that one graph is just one graph. A single graph isn't enough to prove any kind of universal phenomenon. What we see here applies to Methanococcus maripaludis, an Archaeal anaerobe that may or may not share similarities (vis-a-vis DNA repair) with other organisms.
Sunday, July 14, 2013
DNA Strand Asymmetry: More Surprises
The surprises just keep coming. When you start doing comparative genomics on the desktop (which is so easy with all the great tools at genomevolution.org and elsewhere), it's amazing how quickly you run into things that make you slap yourself on the side of the head and go "Whaaaa????"
If you know anything about DNA (or even if you don't), this one will set you back.
I've written before about Chargaff's second parity rule, which (peculiarly) states that A = T and G = C not just for double-stranded DNA (that's the first parity rule) but for bases in a single strand of DNA. The first parity rule is basic: It's what allows one strand of DNA to be complementary to another. The second parity rule is not so intuitive. Why should the amount of adenine have to equal the amount of thymine (or guanine equal cytosine) in a single strand of DNA? The conventional argument is that nature doesn't play favorites with purines and pyrimidines. There's no reason (in theory) why a single strand of DNA should have an excess of purines over pyrimidines or vice versa, all things being equal.
But it turns out, strand asymmetry vis-a-vis purines and pyrimidines is not only not uncommon, it's the rule. (Some call it Szybalski's rule, in fact.) You can prove it to yourself very easily. If you obtain a codon usage chart for a particular organism, then add the frequencies of occurrence of each base in each codon, you can get the relative abundances of the four bases (A, G, T, C) for the coding regions on which the codon chart was based. Let's take a simple example that requires no calculation: Clostridium botulinum. Just by eyeballing the chart below, you can quickly see that (for C. botulinum) codons using purines A and G are way-more-often used than codons containing pyrimidines T and C. (Note the green-highlighted codons.)

If you do the math, you'll find that in C. botulinum, G and A (combined) outnumber T and C by a factor of 1.41. That's a pretty extreme purine:pyrimidine ratio. (Remember that we're dealing with a single strand of DNA here. Codon frequencies are derived from the so-called "message strand" of DNA in coding regions.)
I've done this calculation for 1,373 different bacterial species (don't worry, it's all automated), and the bottom line is, the greater the DNA's A+T content (or, equivalently, the less its G+C content), the greater the purine imbalance. (See this post for a nice graph.)
If you inspect enough codon charts you'll quickly realize that Chargaff's second parity rule never holds true (except now and then by chance). It's a bogus rule, at least in coding regions (DNA that actually gets transcribed in vivo). It may have applicability to pseudogenes or "junk DNA" (but then again, I haven't checked; it may well not apply there either).
If Chargaff's second rule were true, we would expect to find that G = C (and A = T), because that's what the rule says. I went through the codon frequency data for 1,373 different bacterial species and then plotted the ratio of G to C (which Chargaff says should equal 1.0) for each species against the A+T content (which is a kind of phylogenetic signature) for each species. I was shocked by what I found:
I wasn't so much shocked by the fact that Chargaff's rule doesn't hold; I already knew that. What's shocking is that the ratio of G to C goes up as A+T increases, which means G/C is going up even as G+C is going down. (By definition, G+C goes down as A+T goes up.)
Chargaff says G/C should always equal 1.0. In reality, it never does except by chance. What we find is, the less G (or C) the DNA has, the greater the ratio of G to C. To put it differently: At the high-AT end of the phylogenetic scale, cytosine is decreasing faster (much faster) than guanine, as overall G+C content goes down.
When I first plotted this graph, I used a linear regression to get a line that minimizes the sum of squared absolute error. That line turned out to be given by 0.638 + [A+T]. Then I saw that the data looked exponential, not linear. So I refitted the data with a power curve (the red curve shown above) given by
which fit the data even better (minimum summed error 0.1119 instead of 0.1197). What struck me as strange is that the Golden Ratio (1.618) shows up in the power-curve formula (above), but also, the linear form of the regression has G/C equaliing 1.638 when [A+T] goes to 1.0. Which is almost the Golden Ratio.
In a previous post, I mentioned finding that the ratio A/T tends to approximate the Golden Ratio as A+T approaches 1.0. If this were to hold true, it could mean that A/T and G/C both approach the Golden Ratio as A+T approaches 1.0, which would be weird indeed.
For now, I'm not going to make the claim that the Golden Ratio figures into any of this, because it reeks too much of numerology and Intelligent Design (and I'm a fan of neither). I do think it's mildly interesting that A/T and G/C both approach a similar number as A+T approaches unity.
Comments, as usual, are welcome.
If you know anything about DNA (or even if you don't), this one will set you back.
I've written before about Chargaff's second parity rule, which (peculiarly) states that A = T and G = C not just for double-stranded DNA (that's the first parity rule) but for bases in a single strand of DNA. The first parity rule is basic: It's what allows one strand of DNA to be complementary to another. The second parity rule is not so intuitive. Why should the amount of adenine have to equal the amount of thymine (or guanine equal cytosine) in a single strand of DNA? The conventional argument is that nature doesn't play favorites with purines and pyrimidines. There's no reason (in theory) why a single strand of DNA should have an excess of purines over pyrimidines or vice versa, all things being equal.
But it turns out, strand asymmetry vis-a-vis purines and pyrimidines is not only not uncommon, it's the rule. (Some call it Szybalski's rule, in fact.) You can prove it to yourself very easily. If you obtain a codon usage chart for a particular organism, then add the frequencies of occurrence of each base in each codon, you can get the relative abundances of the four bases (A, G, T, C) for the coding regions on which the codon chart was based. Let's take a simple example that requires no calculation: Clostridium botulinum. Just by eyeballing the chart below, you can quickly see that (for C. botulinum) codons using purines A and G are way-more-often used than codons containing pyrimidines T and C. (Note the green-highlighted codons.)

If you do the math, you'll find that in C. botulinum, G and A (combined) outnumber T and C by a factor of 1.41. That's a pretty extreme purine:pyrimidine ratio. (Remember that we're dealing with a single strand of DNA here. Codon frequencies are derived from the so-called "message strand" of DNA in coding regions.)
I've done this calculation for 1,373 different bacterial species (don't worry, it's all automated), and the bottom line is, the greater the DNA's A+T content (or, equivalently, the less its G+C content), the greater the purine imbalance. (See this post for a nice graph.)
If you inspect enough codon charts you'll quickly realize that Chargaff's second parity rule never holds true (except now and then by chance). It's a bogus rule, at least in coding regions (DNA that actually gets transcribed in vivo). It may have applicability to pseudogenes or "junk DNA" (but then again, I haven't checked; it may well not apply there either).
If Chargaff's second rule were true, we would expect to find that G = C (and A = T), because that's what the rule says. I went through the codon frequency data for 1,373 different bacterial species and then plotted the ratio of G to C (which Chargaff says should equal 1.0) for each species against the A+T content (which is a kind of phylogenetic signature) for each species. I was shocked by what I found:
 |
| Using base abundances derived from codon frequency data, I calculated G/C for 1,373 bacterial species and plotted it against total A+T content. (Each dot represents a genome for a particular organism.) Chargaff's second parity rule predicts a horizontal line at y=1.0. Clearly, that rule doesn't hold. |
I wasn't so much shocked by the fact that Chargaff's rule doesn't hold; I already knew that. What's shocking is that the ratio of G to C goes up as A+T increases, which means G/C is going up even as G+C is going down. (By definition, G+C goes down as A+T goes up.)
Chargaff says G/C should always equal 1.0. In reality, it never does except by chance. What we find is, the less G (or C) the DNA has, the greater the ratio of G to C. To put it differently: At the high-AT end of the phylogenetic scale, cytosine is decreasing faster (much faster) than guanine, as overall G+C content goes down.
When I first plotted this graph, I used a linear regression to get a line that minimizes the sum of squared absolute error. That line turned out to be given by 0.638 + [A+T]. Then I saw that the data looked exponential, not linear. So I refitted the data with a power curve (the red curve shown above) given by
G/C = 1.0 + 0.587*[A+T] + 1.618*[A+T]2
which fit the data even better (minimum summed error 0.1119 instead of 0.1197). What struck me as strange is that the Golden Ratio (1.618) shows up in the power-curve formula (above), but also, the linear form of the regression has G/C equaliing 1.638 when [A+T] goes to 1.0. Which is almost the Golden Ratio.
In a previous post, I mentioned finding that the ratio A/T tends to approximate the Golden Ratio as A+T approaches 1.0. If this were to hold true, it could mean that A/T and G/C both approach the Golden Ratio as A+T approaches 1.0, which would be weird indeed.
For now, I'm not going to make the claim that the Golden Ratio figures into any of this, because it reeks too much of numerology and Intelligent Design (and I'm a fan of neither). I do think it's mildly interesting that A/T and G/C both approach a similar number as A+T approaches unity.
Comments, as usual, are welcome.
Friday, July 12, 2013
Do-It-Yourself Phylogenetic Trees
I've been doing a lot of desktop science lately, and I'm happy to report that superb, easy-to-use online tools exist for creating your own phylogenetic trees based on gene similarities, something that's non-trivial to implement yourself.
The other day, I speculated that the fruit-fly Ogg1 gene, which encodes an enzyme designed to repair oxidatively damaged guanine residues in DNA, might derive from Archaea. The Archaea (in case you're not a microbiologist) comprise one of three super-kingdoms in the tree of life. Basically, all life on earth can be classified as either Archaeal, Eukaryotic, or Eubacterial. The Eubacteria are "true bacteria": they're what you and I think of when we think "bacteria." (So, think Staphylococcus and tetanus bacteria and E. coli and all the rest.) The Eukaryota are higher life forms, starting with yeast and fungi and algae and plankton, progressing up through grass and corn and pine trees, worms and rabbits and donkeys, all the way to the highest life form of all, Stephen Colbert. (A little joke there.) Eukaryotes have big, complex cells with a distinct nucleus, complex organelles (like mitochondria and chloroplasts), and a huge amount of DNA packaged into pairs of chromosomes.
Archaea look a lot like bacteria (they're tiny and lack a distinct nucleus, organelles, etc.), and were in fact considered bacteria until recently. But around the turn of the 21st century, Carl Woese and George E. Fox provided persuasive evidence that members of this group of organisms were so different in genetic profile (not to mention lifestyle) that they deserved their own taxonomic domain. Thus, we now recognize certain bacteria-like creatures as Archaea.
The technical considerations behind the distinction between bacteria and archeons are rather deep and have to do with codon usage patterns, ribosomal RNA structure, cell-wall details, lipid metabolism, and other esoterica, but one distinguishing feature of archeons that's easy to understand is their willingness to live under harsh conditions. Archaeal species tend to be what we call extremophiles: They usually (not always) take up residence in places that are incredibly salty, or incredibly hot, or incredibly alkaline or acidic.
While it's generally agreed that eukaryotes arose after Archaea and bacteria appeared, it's by no means clear whether Archaea and bacteria branched off independently from a common ancestor, or perhaps one arose from the other. (A popular theory right now is that Archaea arose from gram-positive bacteria and sought refuge in inhospitable habitats to escape the chemical-warfare tactics of the gram-positives.) A complication that makes studying this sort of thing harder is the fact that horizontal gene transfer has been known to happen (with surprising frequency, actually) across domains.
Is it possible to study phylogenetic relationships, yourself, on the desktop? Of course. One way to do it: Obtain the DNA sequences of a given gene as produced by a variety of organisms, then feed those gene sequences to a tool like the tree-making tool at http://www.phylogeny.fr. Voila! Instant phylogeny.
The Ogg1 gene is an interesting case, because although the DNA-repair enzyme encoded by this gene occurs in a wide variety of higher life forms, plus Archaea, it is not widespread among bacteria. Aside from a couple of Spirochaetes and one Bacteroides species, the only bacteria that have this particular gene are the members of class Clostridia (which are all strict anaerobes). Question: Did the Clostridia get this gene from anaerobic Archaea?
Using the excellent online CoGeBlast tool, I was able to build a list of organisms that have Ogg1 and obtain the relevant gene sequences, all with literally just a few mouse clicks. Once you run a search using CoGeBlast, you can check the checkboxes next to organisms in the results list, then select "Phylogenetics" from the dropdown menu at the bottom of the results list. (See screenshot.)

When you click the Go button, a new FastaView window will open up, containing the gene sequences of all the items whose checkboxes you checked in CoGeBlast. At the bottom of this FastaView window, there's a small box that looks like this:

Click Phylogeny.fr button (red arrow). Immediately, your sequences are sent to the French server where they'll be converted to a phylogenetic tree in a matter of one to two minutes (usually). The result is a tree that looks something like this:

I've color-coded this tree to make the results easier to interpret. Creating a tree of this kind is not without potential pitfalls, because for one thing, if your DNA sequences are of vastly unequal lengths, the groupings made by Phylogeny.fr are likely to reflect gene lengths more than true phylogeny. For this tree, I did various data checks to make sure we're comparing apples and apples. Even so, a sanity check is in order. Do the groupings make sense? They do, actually. At the very top of the diagram (color-coded in green) we find all the eukaryotes grouped together: fruit-fly (Drosophila), yeast (Saccharomyces), fungus (Aspergillus). At the bottom of the diagram, Clostridium species (purplish red) fall into a subtree of their own, next to a tiny subtree of Methoanobrevibacter. This actually makes a good deal of sense, because the two Methanobrevibacter species shown are inhabitants of feces, as are the nearby Clostridium bartletti and C. diff. The fact that all the salt-loving Archaea members group together (organisms with names starting with 'H') is also indicative of a sound grouping. Overall, the tree looks sound.
If you're wondering what all the numbers are, the scale bar at the bottom (0.4) shows the approximate percentage difference in DNA sequences associated with that particular length of tree depth. The red numbers on the tree branches are indicative of the probability that the immediately underlying nodes are related. Probably the most important thing to know is that the evolutionary distance between any two leaves in the tree is proportional to the sums of the branch lengths connecting them. (The branch lengths are not explicitly specified; you have to eyeball it.) At the top of the diagram, you can see that the branch lengths of the two Drosophila instances are very short. This means they're closely related. By contrast, the branch lengths for Saccharomyces and the ancestor to Drosophila are long, meaning that these organisms are distantly related.
Just to give you an idea of the relatedness, I checked the C. botulinum Ogg1 protein amino-acid sequence against C. tetani, and found 63% identity of amino acids. When I compared C. botulinum's enzyme against C. difficile's, there was 52% identity. With Drosophila there is only 32% identity, and even that applies only to a 46% coverage area (versus 90%+ for C. tetani and C. diff). Bottom line, the Blast-wise relatedness does appear to correspond, in sound fashion, to tree-wise relatedness.
Two things stand out. One is that not all of the Clostridium species group together. (There's a small cluster of Clostridia near the salt-lovers, then a main branch near the methane-producing Archaea. The out-group of Clostridia near the salt-lovers happen to all have chromosomal G+C content of 50% or more, which makes them quite different from the rest of the Clositridia, whose G+C is under 30%.) The other thing that stands out is that it does appear as if Clostridial Ogg1 could be Archaeal in origin, based on the relationship of Methanoplanus and Methanobrevibacter to the main group of Clostridia. (Also, the C. leptum group's Ogg1 may share an ancestor with the halophilic Archaea.) One thing we can say for sure is that Ogg1 is ancient.
It's tempting to speculate that the eukaryotes obtained Ogg1 from early mitochondria, and that early mitochondria were actually Archaeal endosymbionts. The first part is easily true, because we know that early mitochondria quickly exported most of their DNA to the host nucleus. (Today's mitochondrial DNA is vestigial. Well over 90% of mitochondrial genes are actually in the host nucleus. Things like mitochondrial DNA polymerase have to be transcribed from nucleus-generated RNA.) Whether or not early mitochondria were Archaeal endosymbionts, no one knows.
Anyway, I hope this shows how easy it is to generate phylogenetic trees from the comfort of a living room sofa, using nothing more than a laptop with wireless internet connection. Try making your own phylo-trees using CoGeBlast and Phylogeny.fr—and let me know what you find out.
The other day, I speculated that the fruit-fly Ogg1 gene, which encodes an enzyme designed to repair oxidatively damaged guanine residues in DNA, might derive from Archaea. The Archaea (in case you're not a microbiologist) comprise one of three super-kingdoms in the tree of life. Basically, all life on earth can be classified as either Archaeal, Eukaryotic, or Eubacterial. The Eubacteria are "true bacteria": they're what you and I think of when we think "bacteria." (So, think Staphylococcus and tetanus bacteria and E. coli and all the rest.) The Eukaryota are higher life forms, starting with yeast and fungi and algae and plankton, progressing up through grass and corn and pine trees, worms and rabbits and donkeys, all the way to the highest life form of all, Stephen Colbert. (A little joke there.) Eukaryotes have big, complex cells with a distinct nucleus, complex organelles (like mitochondria and chloroplasts), and a huge amount of DNA packaged into pairs of chromosomes.
Archaea look a lot like bacteria (they're tiny and lack a distinct nucleus, organelles, etc.), and were in fact considered bacteria until recently. But around the turn of the 21st century, Carl Woese and George E. Fox provided persuasive evidence that members of this group of organisms were so different in genetic profile (not to mention lifestyle) that they deserved their own taxonomic domain. Thus, we now recognize certain bacteria-like creatures as Archaea.
The technical considerations behind the distinction between bacteria and archeons are rather deep and have to do with codon usage patterns, ribosomal RNA structure, cell-wall details, lipid metabolism, and other esoterica, but one distinguishing feature of archeons that's easy to understand is their willingness to live under harsh conditions. Archaeal species tend to be what we call extremophiles: They usually (not always) take up residence in places that are incredibly salty, or incredibly hot, or incredibly alkaline or acidic.
While it's generally agreed that eukaryotes arose after Archaea and bacteria appeared, it's by no means clear whether Archaea and bacteria branched off independently from a common ancestor, or perhaps one arose from the other. (A popular theory right now is that Archaea arose from gram-positive bacteria and sought refuge in inhospitable habitats to escape the chemical-warfare tactics of the gram-positives.) A complication that makes studying this sort of thing harder is the fact that horizontal gene transfer has been known to happen (with surprising frequency, actually) across domains.
Is it possible to study phylogenetic relationships, yourself, on the desktop? Of course. One way to do it: Obtain the DNA sequences of a given gene as produced by a variety of organisms, then feed those gene sequences to a tool like the tree-making tool at http://www.phylogeny.fr. Voila! Instant phylogeny.
The Ogg1 gene is an interesting case, because although the DNA-repair enzyme encoded by this gene occurs in a wide variety of higher life forms, plus Archaea, it is not widespread among bacteria. Aside from a couple of Spirochaetes and one Bacteroides species, the only bacteria that have this particular gene are the members of class Clostridia (which are all strict anaerobes). Question: Did the Clostridia get this gene from anaerobic Archaea?
Using the excellent online CoGeBlast tool, I was able to build a list of organisms that have Ogg1 and obtain the relevant gene sequences, all with literally just a few mouse clicks. Once you run a search using CoGeBlast, you can check the checkboxes next to organisms in the results list, then select "Phylogenetics" from the dropdown menu at the bottom of the results list. (See screenshot.)

When you click the Go button, a new FastaView window will open up, containing the gene sequences of all the items whose checkboxes you checked in CoGeBlast. At the bottom of this FastaView window, there's a small box that looks like this:

Click Phylogeny.fr button (red arrow). Immediately, your sequences are sent to the French server where they'll be converted to a phylogenetic tree in a matter of one to two minutes (usually). The result is a tree that looks something like this:

I've color-coded this tree to make the results easier to interpret. Creating a tree of this kind is not without potential pitfalls, because for one thing, if your DNA sequences are of vastly unequal lengths, the groupings made by Phylogeny.fr are likely to reflect gene lengths more than true phylogeny. For this tree, I did various data checks to make sure we're comparing apples and apples. Even so, a sanity check is in order. Do the groupings make sense? They do, actually. At the very top of the diagram (color-coded in green) we find all the eukaryotes grouped together: fruit-fly (Drosophila), yeast (Saccharomyces), fungus (Aspergillus). At the bottom of the diagram, Clostridium species (purplish red) fall into a subtree of their own, next to a tiny subtree of Methoanobrevibacter. This actually makes a good deal of sense, because the two Methanobrevibacter species shown are inhabitants of feces, as are the nearby Clostridium bartletti and C. diff. The fact that all the salt-loving Archaea members group together (organisms with names starting with 'H') is also indicative of a sound grouping. Overall, the tree looks sound.
If you're wondering what all the numbers are, the scale bar at the bottom (0.4) shows the approximate percentage difference in DNA sequences associated with that particular length of tree depth. The red numbers on the tree branches are indicative of the probability that the immediately underlying nodes are related. Probably the most important thing to know is that the evolutionary distance between any two leaves in the tree is proportional to the sums of the branch lengths connecting them. (The branch lengths are not explicitly specified; you have to eyeball it.) At the top of the diagram, you can see that the branch lengths of the two Drosophila instances are very short. This means they're closely related. By contrast, the branch lengths for Saccharomyces and the ancestor to Drosophila are long, meaning that these organisms are distantly related.
Just to give you an idea of the relatedness, I checked the C. botulinum Ogg1 protein amino-acid sequence against C. tetani, and found 63% identity of amino acids. When I compared C. botulinum's enzyme against C. difficile's, there was 52% identity. With Drosophila there is only 32% identity, and even that applies only to a 46% coverage area (versus 90%+ for C. tetani and C. diff). Bottom line, the Blast-wise relatedness does appear to correspond, in sound fashion, to tree-wise relatedness.
Two things stand out. One is that not all of the Clostridium species group together. (There's a small cluster of Clostridia near the salt-lovers, then a main branch near the methane-producing Archaea. The out-group of Clostridia near the salt-lovers happen to all have chromosomal G+C content of 50% or more, which makes them quite different from the rest of the Clositridia, whose G+C is under 30%.) The other thing that stands out is that it does appear as if Clostridial Ogg1 could be Archaeal in origin, based on the relationship of Methanoplanus and Methanobrevibacter to the main group of Clostridia. (Also, the C. leptum group's Ogg1 may share an ancestor with the halophilic Archaea.) One thing we can say for sure is that Ogg1 is ancient.
It's tempting to speculate that the eukaryotes obtained Ogg1 from early mitochondria, and that early mitochondria were actually Archaeal endosymbionts. The first part is easily true, because we know that early mitochondria quickly exported most of their DNA to the host nucleus. (Today's mitochondrial DNA is vestigial. Well over 90% of mitochondrial genes are actually in the host nucleus. Things like mitochondrial DNA polymerase have to be transcribed from nucleus-generated RNA.) Whether or not early mitochondria were Archaeal endosymbionts, no one knows.
Anyway, I hope this shows how easy it is to generate phylogenetic trees from the comfort of a living room sofa, using nothing more than a laptop with wireless internet connection. Try making your own phylo-trees using CoGeBlast and Phylogeny.fr—and let me know what you find out.
Wednesday, July 10, 2013
Shedding Light on DNA Strand Asymmetry
In 1950, Erwin Chargaff was the first to report that the amount of adenine (A) in DNA equals the amount of thymine (T), and the amount of guanine (G) equals the amount of cytosine (C). This result was instrumental in helping Watson and Crick (and Rosalind Franklin) determine the structure of DNA.
It's pretty easy to understand that every A on one strand of DNA pairs with a T on the other strand (and every G pairs with an opposite-strand C); this explains DNA complementarity and the associated replication model. But somewhere along the line, Chargaff was credited with the much less obvious rule that A = T and G = C even for individual strands of DNA that aren't paired with anything. This is the so-called second parity rule attributed to Chargaff, although I can't find any record of Chargaff himself having postulated such a rule. The Chargaff papers that are so often cited as supporting this rule (in particular the 3-paper series culminating in this report in PNAS) do not, in fact, offer such a rule, and if you read the papers carefully, what Chargaff and colleagues actually found was that one strand of DNA is heavier than the other (they label the strands 'H' and 'L', for Heavy and Light); not only that, but Chargaff et al. reported a consistent difference in purine content between strands (see Table 1 of this paper).
When I interviewed Linus Pauling in 1977, he cautioned me to always read the Results section of a paper carefully, because people will often conclude something entirely different than what the Results actually showed, or cite a paper as showing "ABC" when the data actually showed "XYZ."
How right he was.
At any rate, it turns out that the "message" strand of a gene hardly ever contains equal amounts of purines and pyrimidines. Codon analysis reveals that as genes become richer in A+T content (or as G+C content goes down), the excess of purines on the message strand becomes larger and larger. This is depicted in the following graph, which shows message-strand purine content (A+G) plotted against A+T content, for 1,373 distinct bacterial species. (No species is represented twice.)
It's quite obvious that when A+T content is above approximately 33%, as it is for most bacterial species, the message strand tends to be comparatively purine-rich. Below A+T = 33%, the message strand becomes more pyrimidine-rich than purine-rich. (Note: In bacteria, where most of the DNA is in coding regions, codon-derived A+T content is very close to whole-genome A+T content. I checked the 1,373 species graphed here and found whole-chromosome A+T to differ from codon-derived A+T by an average of less than 7 parts in 10,000.)
The correlation between A+T and purine content is strong (r=0.85). Still, you can see that quite a few points have drifted far from the regression line, especially in the region of x = 0.5 to x = 0.7, where lots of points lie above y = 0.55. What's going on with those organisms? I decided to do some investigating.
First, some basics. Over time, transition mutations (AT↔GC) can change an organism's A+T content and thus move it along the x-axis of the graph, but transitions cannot move an organism higher or lower on the graph, because (by definition) transitions don't affect the strandwise purine balance.
Transversions, on the other hand, can affect strandwise purine balance (in theory, at least), but only if they occur more often on one strand of DNA than the other. (I should say: occur more often, or are fixed more often, on one strand versus the other.) For example, let's say G-to-T transversions are the most common kind of transversion (which is probably true, given that guanine is the most easily oxidized of the four bases and given the fact that failure to repair 8-oxoguanine lesions does lead to eventual replacement with thymine). And let's say G-to-T transversions are most likely to occur on the non-transcribed strand of DNA, at transcription time. (The non-transcribed strand is uncoiled and unprotected while transcription is taking place on the other strand.) Over time, the non-transcribed strand would lose guanines; they'd be replaced by thymines. The message strand, or RNA-synonymous strand (which is also the non-transcribed strand) would become pyrimidine-rich and the other strand would become purine-rich.
Unfortunately, while that's exactly what happens for organisms with A+T content below 33%, precisely the opposite happens (purines accumulate on the message strand) in organisms with A+T above 33%. And in fact, in some high-AT organisms, the purine content of message strands is rather extreme. How can we explain that?
One possibility is that some organisms have evolved extremely effective transversion repair systems for the message (non-transcribed) strand of genes—systems that are so effective, no G-to-T transversions go unrepaired on the message strand. The transcribed strand, on the other hand, doesn't get the benefit of this repair system, possibly because the repair enzymes can't access the strand: it's engulfed in transcription factors, topoisomerases, RNA polymerase, nearby ribosomal machinery, etc.
If the non-transcribed strand never mutates (because all mutations are swiftly repaired), then the transcribed strand will (in the absence of equally effective repairs) eventually accumulate G-to-T mutations, and the message strand will accumulate adenines (purines). Perhaps.
In the graph further above, you'll notice at x = 0.6 a tiny spur of points hangs down at around y = 0.5. These points belong to some Bartonella species, plus a Parachlamydia and another chlamydial organism. These are endosymbionts that have lost a good portion of their genomes over time. It seems likely they've lost some transversion-repair machinery. During transcription, their message strands are going unrepaired. G-to-T transversions happen on the message strand, rendering it light in purines. Such a scenario seems plausible, at least.
By this reasoning, maybe points far above the regression line represent organisms that have gained repair functionality, such that their message strands never undergo G-to-T transversions (although their transcribed strands do). Is this possible?
Examination of the highest points on the graph shows a predominance of Clostridia. (Not just members of the genus Clostridium, but the class Clostridia, which is a large, ancient, and diverse class of anaerobes.) One thing we know about the Clostridia is that unlike all other bacteria (unlike members of the Gammaproteobacteria, the Alpha- and Betaproteobacteria, the Actinomycetes, the Bacteroidetes, etc.), the Clostridia have Ogg1, otherwise known as 8-oxoguanine glycosylase (which specifically prevents G-to-T transversions). They share this capability with all members of the Archaea, and all higher life forms as well.
Note that while non-Ogg1 enzymes exist for correcting 8-oxoguanine lesions (e.g., MutM, MutY, mfd), there is evidence that Ogg1 is specifically involved in repair of 8oxoG lesions in non-transcribed strands of DNA, at transcription time. (The other 8oxoG repair systems may not be strand-specific.)
If Archaea benefit from Ogg1 the way Clostridia do, they too should fall well above the regression line on a graph of A+G versus A+T. And this is exactly what we find. In the graph below, the pink squares are members of Archaea that came up positive in a protein-Blast query against Drosophila Ogg1. (I'll explain why I used Drosophila in a minute.) The red-orange circles are bacterial species (mostly from class Clostridia) that turned up Ogg1-positive in a similar Blast search.
The points in this plot are significantly higher on the y-axis than points in the all-bacteria plot (and the regression line is steeper), consistent with a different DNA repair profile.
In identifying Ogg1-positive organisms, I wanted to avoid false positives (organisms with enzymes that share characteristics of Ogg1 but that aren't truly Ogg1), so for the Blast query I used Drosophila's Ogg1 as a reference enzyme, since it is well studied (unlike Archaeal or Clostridial Ogg1). I also set the E-value cutoff at 1e-10, to reduce spurious matches with DNA repair enzymes or nucleases that might have domain similarity with Ogg1 but aren't Ogg1. In addition, I did spot checks to be sure the putative Ogg1 matches that came up were not actually matches of Fpg (MutM), RecA, RadA, MutY, DNA-3-methyladenine glycosidase, or other DNA-binding enzymes.
Bottom line, organisms that have an Archaeal 8-oxoguanine glycosylase enzyme (mostly obligate anaerobes) occupy a unique part of the A+G vs. A+T graph. Which makes sense. It's only logical that anaerobes would have different DNA repair strategies (and a different "repairosome") than oxygen-tolerant bacteria, because oxidative stress is, in general, handled much differently in anaerobes. The fact that they bring different repair tactics to bear on DNA shouldn't come as a surprise.
It's pretty easy to understand that every A on one strand of DNA pairs with a T on the other strand (and every G pairs with an opposite-strand C); this explains DNA complementarity and the associated replication model. But somewhere along the line, Chargaff was credited with the much less obvious rule that A = T and G = C even for individual strands of DNA that aren't paired with anything. This is the so-called second parity rule attributed to Chargaff, although I can't find any record of Chargaff himself having postulated such a rule. The Chargaff papers that are so often cited as supporting this rule (in particular the 3-paper series culminating in this report in PNAS) do not, in fact, offer such a rule, and if you read the papers carefully, what Chargaff and colleagues actually found was that one strand of DNA is heavier than the other (they label the strands 'H' and 'L', for Heavy and Light); not only that, but Chargaff et al. reported a consistent difference in purine content between strands (see Table 1 of this paper).
When I interviewed Linus Pauling in 1977, he cautioned me to always read the Results section of a paper carefully, because people will often conclude something entirely different than what the Results actually showed, or cite a paper as showing "ABC" when the data actually showed "XYZ."
How right he was.
At any rate, it turns out that the "message" strand of a gene hardly ever contains equal amounts of purines and pyrimidines. Codon analysis reveals that as genes become richer in A+T content (or as G+C content goes down), the excess of purines on the message strand becomes larger and larger. This is depicted in the following graph, which shows message-strand purine content (A+G) plotted against A+T content, for 1,373 distinct bacterial species. (No species is represented twice.)
 |
| Codon analysis reveals that as A+T content increases, message-strand purine content (A+G) increases. Each point on this graph represents a unique bacterial species (N=1373). |
It's quite obvious that when A+T content is above approximately 33%, as it is for most bacterial species, the message strand tends to be comparatively purine-rich. Below A+T = 33%, the message strand becomes more pyrimidine-rich than purine-rich. (Note: In bacteria, where most of the DNA is in coding regions, codon-derived A+T content is very close to whole-genome A+T content. I checked the 1,373 species graphed here and found whole-chromosome A+T to differ from codon-derived A+T by an average of less than 7 parts in 10,000.)
The correlation between A+T and purine content is strong (r=0.85). Still, you can see that quite a few points have drifted far from the regression line, especially in the region of x = 0.5 to x = 0.7, where lots of points lie above y = 0.55. What's going on with those organisms? I decided to do some investigating.
First, some basics. Over time, transition mutations (AT↔GC) can change an organism's A+T content and thus move it along the x-axis of the graph, but transitions cannot move an organism higher or lower on the graph, because (by definition) transitions don't affect the strandwise purine balance.
Transversions, on the other hand, can affect strandwise purine balance (in theory, at least), but only if they occur more often on one strand of DNA than the other. (I should say: occur more often, or are fixed more often, on one strand versus the other.) For example, let's say G-to-T transversions are the most common kind of transversion (which is probably true, given that guanine is the most easily oxidized of the four bases and given the fact that failure to repair 8-oxoguanine lesions does lead to eventual replacement with thymine). And let's say G-to-T transversions are most likely to occur on the non-transcribed strand of DNA, at transcription time. (The non-transcribed strand is uncoiled and unprotected while transcription is taking place on the other strand.) Over time, the non-transcribed strand would lose guanines; they'd be replaced by thymines. The message strand, or RNA-synonymous strand (which is also the non-transcribed strand) would become pyrimidine-rich and the other strand would become purine-rich.
Unfortunately, while that's exactly what happens for organisms with A+T content below 33%, precisely the opposite happens (purines accumulate on the message strand) in organisms with A+T above 33%. And in fact, in some high-AT organisms, the purine content of message strands is rather extreme. How can we explain that?
One possibility is that some organisms have evolved extremely effective transversion repair systems for the message (non-transcribed) strand of genes—systems that are so effective, no G-to-T transversions go unrepaired on the message strand. The transcribed strand, on the other hand, doesn't get the benefit of this repair system, possibly because the repair enzymes can't access the strand: it's engulfed in transcription factors, topoisomerases, RNA polymerase, nearby ribosomal machinery, etc.
If the non-transcribed strand never mutates (because all mutations are swiftly repaired), then the transcribed strand will (in the absence of equally effective repairs) eventually accumulate G-to-T mutations, and the message strand will accumulate adenines (purines). Perhaps.
In the graph further above, you'll notice at x = 0.6 a tiny spur of points hangs down at around y = 0.5. These points belong to some Bartonella species, plus a Parachlamydia and another chlamydial organism. These are endosymbionts that have lost a good portion of their genomes over time. It seems likely they've lost some transversion-repair machinery. During transcription, their message strands are going unrepaired. G-to-T transversions happen on the message strand, rendering it light in purines. Such a scenario seems plausible, at least.
By this reasoning, maybe points far above the regression line represent organisms that have gained repair functionality, such that their message strands never undergo G-to-T transversions (although their transcribed strands do). Is this possible?
Examination of the highest points on the graph shows a predominance of Clostridia. (Not just members of the genus Clostridium, but the class Clostridia, which is a large, ancient, and diverse class of anaerobes.) One thing we know about the Clostridia is that unlike all other bacteria (unlike members of the Gammaproteobacteria, the Alpha- and Betaproteobacteria, the Actinomycetes, the Bacteroidetes, etc.), the Clostridia have Ogg1, otherwise known as 8-oxoguanine glycosylase (which specifically prevents G-to-T transversions). They share this capability with all members of the Archaea, and all higher life forms as well.
Note that while non-Ogg1 enzymes exist for correcting 8-oxoguanine lesions (e.g., MutM, MutY, mfd), there is evidence that Ogg1 is specifically involved in repair of 8oxoG lesions in non-transcribed strands of DNA, at transcription time. (The other 8oxoG repair systems may not be strand-specific.)
If Archaea benefit from Ogg1 the way Clostridia do, they too should fall well above the regression line on a graph of A+G versus A+T. And this is exactly what we find. In the graph below, the pink squares are members of Archaea that came up positive in a protein-Blast query against Drosophila Ogg1. (I'll explain why I used Drosophila in a minute.) The red-orange circles are bacterial species (mostly from class Clostridia) that turned up Ogg1-positive in a similar Blast search.
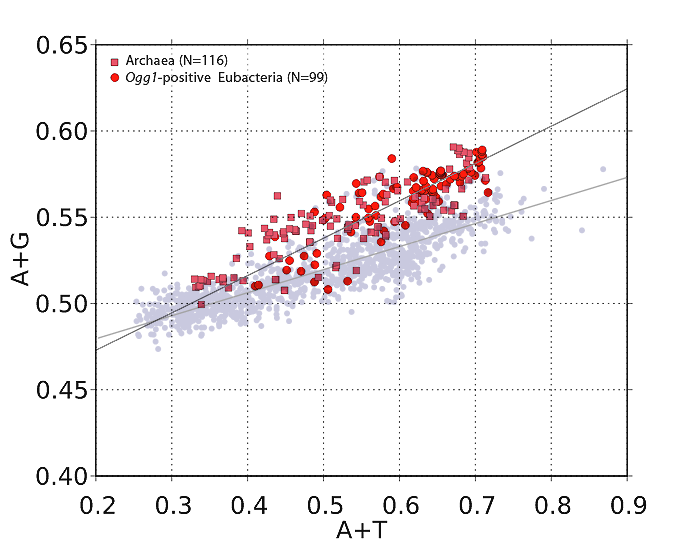 |
| Ogg1-positive organisms are plotted here. The pink squares are Archaea species. Red-orange circles are bacterial species that came up Ogg1-positive in a protein Blast search using a Drosophila Ogg1 amino-acid sequence. In the background (greyed out) is the graph of all 1,373 bacterial species, for comparison. Note how the Ogg1-positive organisms have a higher purine (A+G) content than the vast majority of bacteria. |
The points in this plot are significantly higher on the y-axis than points in the all-bacteria plot (and the regression line is steeper), consistent with a different DNA repair profile.
In identifying Ogg1-positive organisms, I wanted to avoid false positives (organisms with enzymes that share characteristics of Ogg1 but that aren't truly Ogg1), so for the Blast query I used Drosophila's Ogg1 as a reference enzyme, since it is well studied (unlike Archaeal or Clostridial Ogg1). I also set the E-value cutoff at 1e-10, to reduce spurious matches with DNA repair enzymes or nucleases that might have domain similarity with Ogg1 but aren't Ogg1. In addition, I did spot checks to be sure the putative Ogg1 matches that came up were not actually matches of Fpg (MutM), RecA, RadA, MutY, DNA-3-methyladenine glycosidase, or other DNA-binding enzymes.
Bottom line, organisms that have an Archaeal 8-oxoguanine glycosylase enzyme (mostly obligate anaerobes) occupy a unique part of the A+G vs. A+T graph. Which makes sense. It's only logical that anaerobes would have different DNA repair strategies (and a different "repairosome") than oxygen-tolerant bacteria, because oxidative stress is, in general, handled much differently in anaerobes. The fact that they bring different repair tactics to bear on DNA shouldn't come as a surprise.
Monday, July 08, 2013
DNA Repair 101
You don't have to be a biologist to know that anything that can damage DNA is potentially harmful, because it can cause mutations (which are, in fact, mostly harmful; very few mutations are beneficial). Fortunately, cells contain dozens of different kinds of repair enzymes, and most DNA damage is repaired quickly. When damage isn't repaired quickly (or properly), you have a mutation.
It's not much of a stretch to say that DNA repair enzymes play a front-and-center role in evolution (or at least the portion of evolution that's driven by mutations). Which is why molecular geneticists tend to pay a lot of attention to DNA repair processes. Anything that can affect the composition of DNA can change the course of evolution.
DNA is remarkably stable, chemically. Nonetheless, it is vulnerable to oxidative attack (by hydroxyl radicals, superoxides, nitric oxide, and other Reactive Oxygenated Species generated in the course of cell metabolism—never mind exogenous poisons).
Of the four bases in DNA—guanine (G), cytosine (C), adenine (A), thymine (T)—guanine is the most susceptible to oxidative attack. When it's exposed to an oxidant, it can form 7,8-dihydro-8-oxoguanine, OG for short. What can happen then is, the OG residue in DNA pivots around its ribosyl bond until the amino group is facing the other way (see diagram), and when that happens, OG can pair up with adenine instead of guanine's usual partner, cytosine.
Rest assured, there are proofreading enzymes that can and will detect such funny business in short order. But if OG isn't detected and replaced with a normal guanine before replication occurs, OG may get paired up with an adenine during replication (and then it'll eventually be swapped out with thymine, adenine's usual partner). That's bad, because what it means is that a G:C pair ended up getting changed to a T:A pair. (The place of the G got taken first by OG and then T. The place of G's opposite-strand partner, C, eventually got taken by A.) In so many words: that's a mutation.
It turns out there's a special enzyme designed to prevent the G↔T funny business we've just been talking about. It's called oxoguanine glycosylase, or Ogg1 for short. You'll sometimes see it called 8-oxoguanine-DNA-glycosylase, and from a capabilities standpoint it's often (wrongly) compared to the Fpg enzyme (formamidopyrimidine-DNA glycosylase), which is not the same as Ogg1 at all.
Just about all higher life forms have an Ogg1 enzyme (which clips OG out of DNA and ensures it gets replaced with a brand-new guanine before any funny business can happen). Surprisingly few bacteria have this enzyme, instead preferring to let the more general-purpose Fpg (MutM) take its place. If you run a Blast search of a reference Ogg1 gene (the Drosophila version works well) against all bacterial genomes, you'll get only a few hundred matches (out of around 10,000 sequenced bacterial genomes), the vast majority belonging to members of the class Clostridia (a truly fearsome group of anaerobic spore-formers containing the botulism germ, the tetanus bacterium, the notorious C. difficile—also known as C. diff—and some other creatures you probably don't want to meet). If you run the same Blast search against Archaea (this is the other major "germ-like" microbial domain, along with true bacteria), you'll get hits against almost every member species of the Archaea. Personally, I think it's likely the Ogg1 enzyme originated with a common ancestor of today's Archaea and Eukaryota, and arrived in Clostridia by lateral gene transfer (not terribly recently, though).
One thing is certain: E. coli does not have Ogg1, nor does Staphylococcus, nor Streptococcus, nor any germ you've ever heard of (other than the aforementioned Clostridia members, plus Archaea). And yet, every yeast and fungus has it, every plant, every fruit fly, every fish, every human—every higher life form. Ironically, only five members of Archaea turned up positive for the Fpg enzyme when I did a check, whereas almost all Eubacteria ("true bacteria") have it, including Clostridia. Bottom line, Clostridia have the best of both worlds: Fpg, plus Ogg1. Belt and suspenders, both.
This is just a tiny intro to the subject of DNA repair, which is a vast subject indeed. For more, see this article, or just start rummaging around in Google Scholar.
It's not much of a stretch to say that DNA repair enzymes play a front-and-center role in evolution (or at least the portion of evolution that's driven by mutations). Which is why molecular geneticists tend to pay a lot of attention to DNA repair processes. Anything that can affect the composition of DNA can change the course of evolution.
DNA is remarkably stable, chemically. Nonetheless, it is vulnerable to oxidative attack (by hydroxyl radicals, superoxides, nitric oxide, and other Reactive Oxygenated Species generated in the course of cell metabolism—never mind exogenous poisons).
Of the four bases in DNA—guanine (G), cytosine (C), adenine (A), thymine (T)—guanine is the most susceptible to oxidative attack. When it's exposed to an oxidant, it can form 7,8-dihydro-8-oxoguanine, OG for short. What can happen then is, the OG residue in DNA pivots around its ribosyl bond until the amino group is facing the other way (see diagram), and when that happens, OG can pair up with adenine instead of guanine's usual partner, cytosine.
 |
| When guanine is oxidized to form 7,8-dihydro-8-oxoguanine, it mispairs with adenine instead of its usual partner, cytosine. |
It turns out there's a special enzyme designed to prevent the G↔T funny business we've just been talking about. It's called oxoguanine glycosylase, or Ogg1 for short. You'll sometimes see it called 8-oxoguanine-DNA-glycosylase, and from a capabilities standpoint it's often (wrongly) compared to the Fpg enzyme (formamidopyrimidine-DNA glycosylase), which is not the same as Ogg1 at all.
Just about all higher life forms have an Ogg1 enzyme (which clips OG out of DNA and ensures it gets replaced with a brand-new guanine before any funny business can happen). Surprisingly few bacteria have this enzyme, instead preferring to let the more general-purpose Fpg (MutM) take its place. If you run a Blast search of a reference Ogg1 gene (the Drosophila version works well) against all bacterial genomes, you'll get only a few hundred matches (out of around 10,000 sequenced bacterial genomes), the vast majority belonging to members of the class Clostridia (a truly fearsome group of anaerobic spore-formers containing the botulism germ, the tetanus bacterium, the notorious C. difficile—also known as C. diff—and some other creatures you probably don't want to meet). If you run the same Blast search against Archaea (this is the other major "germ-like" microbial domain, along with true bacteria), you'll get hits against almost every member species of the Archaea. Personally, I think it's likely the Ogg1 enzyme originated with a common ancestor of today's Archaea and Eukaryota, and arrived in Clostridia by lateral gene transfer (not terribly recently, though).
One thing is certain: E. coli does not have Ogg1, nor does Staphylococcus, nor Streptococcus, nor any germ you've ever heard of (other than the aforementioned Clostridia members, plus Archaea). And yet, every yeast and fungus has it, every plant, every fruit fly, every fish, every human—every higher life form. Ironically, only five members of Archaea turned up positive for the Fpg enzyme when I did a check, whereas almost all Eubacteria ("true bacteria") have it, including Clostridia. Bottom line, Clostridia have the best of both worlds: Fpg, plus Ogg1. Belt and suspenders, both.
This is just a tiny intro to the subject of DNA repair, which is a vast subject indeed. For more, see this article, or just start rummaging around in Google Scholar.
Sunday, July 07, 2013
More Science on the Desktop
If you took Bacteriology 101, you were probably subjected to (maybe even tested on) the standard mythology about anaerobes lacking the enzyme catalase. The standard mythology goes like this: Almost all life forms (from bacteria to dandelions to humans) have a special enzyme called catalase that detoxifies hydrogen peroxide by breaking it down to water and molecular oxygen. The only exception: strict anaerobes (bacteria that cannot live in the presence of oxygen). They seem to lack catalase.
I've written on this subject before, so I won't bore you with a proper debunking of all aspects of the catalase myth here. (For that, see this post.) Right now, I just want to emphasize one point, which is that, contrary to myth, quite a few strict anaerobes do have catalase. I've listed 87 examples by name below. (Scroll down.)
I have to admit, even I was shocked to find there are 87 species of catalase-positive strict anaerobes among the eubacteria. It's about quadruple the number I would have expected.
If you're curious how I came up with a list of 87 catalase-positive anaerobes, here's how. First, I assembled a sizable (N=1373) list of bacteria, unduplicated at the species level. (So in other words, E. coli is listed only once, Staphylococcus aureus is listed only once, etc. No species is listed twice.) I then used the free/online CoGeBlast tool to run two Blast searches: one designed to identify aerobes, and another to identify catalase-positive organisms. In the end, I had all 1,373 organisms tagged as to whether each was aerobic, anaerobic, catalase-positive, or catalase-negative.
It's not as easy as you'd think to identify strict anaerobes. There is no single enzymatic marker that can be used to identify anaerobes reliably (across 1,373 species), as far as I know. I took the opposite approach, tagging as aerobic any organism that produces cytochrome c oxidase and/or NADH dehydrogenase. (These are enzymes involved in classic oxidative phosphorylation of the kind no strict anaerobe participates in.) In particular, I used the following set of amino acid sequences as markers of aerobic respiration (non-biogeeks, scroll down):
>sp|Q6MIR4|NUOB_BDEBA NADH-quinone oxidoreductase subunit B OS=Bdellovibrio bacteriovorus (strain ATCC 15356 / DSM 50701 / NCIB 9529 / HD100) GN=nuoB PE=3 SV=1
MHNEQVQGLVSHDGMTGTQAVDDMSRGFAFTSKLDAIVAWGRKNSLWPMPYGTACCGIEF MSVMGPKYDLARFGAEVARFSPRQADLLVVAGTITEKMAPVIVRIYQQMLEPKYVLSMGA CASSGGFYRAYHVLQGVDKVIPVDVYIPGCPPTPEAVMDGIMALQRMIATNQPRPWKDNW KSPYEQA
>sp|P0ABJ3|CYOC_ECOLI Cytochrome o ubiquinol oxidase subunit 3 OS=Escherichia coli (strain K12) GN=cyoC PE=1 SV=1
MATDTLTHATAHAHEHGHHDAGGTKIFGFWIYLMSDCILFSILFATYAVLVNGTAGGPTG KDIFELPFVLVETFLLLFSSITYGMAAIAMYKNNKSQVISWLALTWLFGAGFIGMEIYEF HHLIVNGMGPDRSGFLSAFFALVGTHGLHVTSGLIWMAVLMVQIARRGLTSTNRTRIMCL SLFWHFLDVVWICVFTVVYLMGAM
>sp|Q9I425|CYOC_PSEAE Cytochrome o ubiquinol oxidase subunit 3 OS=Pseudomonas aeruginosa (strain ATCC 15692 / PAO1 / 1C / PRS 101 / LMG 12228) GN=cyoC PE=3 SV=1
MSTAVLNKHLADAHEVGHDHDHAHDSGGNTVFGFWLYLMTDCVLFASVFATYAVLVHHTA GGPSGKDIFELPYVLVETAILLVSSCTYGLAMLSAHKGAKGQAIAWLGVTFLLGAAFIGM EINEFHHLIAEGFGPSRSAFLSSFFTLVGMHGLHVSAGLLWMLVLMAQIWTRGLTAQNNT RMMCLSLFWHFLDIVWICVFTVVYLMGAL
>tr|Q7VDD9|Q7VDD9_PROMA Cytochrome c oxidase subunit III OS=Prochlorococcus marinus (strain SARG / CCMP1375 / SS120) GN=cyoC PE=3 SV=1
MTTISSVDKKAEELTSQTEEHPDLRLFGLVSFLVADGMTFAGFFAAYLTFKAVNPLLPDA IYELELPLPTLNTILLLVSSATFHRAGKALEAKESEKCQRWLLITAGLGIAFLVSQMFEY FTLPFGLTDNLYASTFYALTGFHGLHVTLGAIMILIVWWQARSPGGRITTENKFPLEAAE LYWHFVDGIWVILFIILYLL
>sp|Q8KS19|CCOP2_PSEST Cbb3-type cytochrome c oxidase subunit CcoP2 OS=Pseudomonas stutzeri GN=ccoP2 PE=1 SV=1
MTSFWSWYVTLLSLGTIAALVWLLLATRKGQRPDSTEETVGHSYDGIEEYDNPLPRWWFM LFVGTVIFALGYLVLYPGLGNWKGILPGYEGGWTQVKEWQREMDKANEQYGPLYAKYAAM PVEEVAKDPQALKMGGRLFASNCSVCHGSDAKGAYGFPNLTDDDWLWGGEPETIKTTILH GRQAVMPGWKDVIGEEGIRNVAGYVRSLSGRDTPEGISVDIEQGQKIFAANCVVCHGPEA KGVTAMGAPNLTDNVWLYGSSFAQIQQTLRYGRNGRMPAQEAILGNDKVHLLAAYVYSLS QQPEQ
>sp|P57542|CYOC_BUCAI Cytochrome o ubiquinol oxidase subunit 3 OS=Buchnera aphidicola subsp. Acyrthosiphon pisum (strain APS) GN=cyoC PE=3 SV=1
MIENKFNNTILNSNSSTHDKISETKKLFGLWIYLMSDCIMFAVLFAVYAIVSSNISINLI SNKIFNLSSILLETFLLLLSSLSCGFVVIAMNQKRIKMIYSFLTITFIFGLIFLLMEVHE FYELIIENFGPDKNAFFSIFFTLVATHGVHIFFGLILILSILYQIKKLGLTNSIRTRILC FSVFWHFLDIIWICVFTFVYLNGAI
>sp|O24958|CCOP_HELPY Cbb3-type cytochrome c oxidase subunit CcoP OS=Helicobacter pylori (strain ATCC 700392 / 26695) GN=ccoP PE=3 SV=1
MDFLNDHINVFGLIAALVILVLTIYESSSLIKEMRDSKSQGELVENGHLIDGIGEFANNV PVGWIASFMCTIVWAFWYFFFGYPLNSFSQIGQYNEEVKAHNQKFEAKWKHLGQKELVDM GQGIFLVHCSQCHGITAEGLHGSAQNLVRWGKEEGIMDTIKHGSKGMDYLAGEMPAMELD EKDAKAIASYVMAELSSVKKTKNPQLIDKGKELFESMGCTGCHGNDGKGLQENQVFAADL TAYGTENFLRNILTHGKKGNIGHMPSFKYKNFSDLQVKALLNLSNR
>sp|P0ABI8|CYOB_ECOLI Ubiquinol oxidase subunit 1 OS=Escherichia coli (strain K12) GN=cyoB PE=1 SV=1
MFGKLSLDAVPFHEPIVMVTIAGIILGGLALVGLITYFGKWTYLWKEWLTSVDHKRLGIM YIIVAIVMLLRGFADAIMMRSQQALASAGEAGFLPPHHYDQIFTAHGVIMIFFVAMPFVI GLMNLVVPLQIGARDVAFPFLNNLSFWFTVVGVILVNVSLGVGEFAQTGWLAYPPLSGIE YSPGVGVDYWIWSLQLSGIGTTLTGINFFVTILKMRAPGMTMFKMPVFTWASLCANVLII ASFPILTVTVALLTLDRYLGTHFFTNDMGGNMMMYINLIWAWGHPEVYILILPVFGVFSE IAATFSRKRLFGYTSLVWATVCITVLSFIVWLHHFFTMGAGANVNAFFGITTMIIAIPTG VKIFNWLFTMYQGRIVFHSAMLWTIGFIVTFSVGGMTGVLLAVPGADFVLHNSLFLIAHF HNVIIGGVVFGCFAGMTYWWPKAFGFKLNETWGKRAFWFWIIGFFVAFMPLYALGFMGMT RRLSQQIDPQFHTMLMIAASGAVLIALGILCLVIQMYVSIRDRDQNRDLTGDPWGGRTLE WATSSPPPFYNFAVVPHVHERDAFWEMKEKGEAYKKPDHYEEIHMPKNSGAGIVIAAFST IFGFAMIWHIWWLAIVGFAGMIITWIVKSFDEDVDYYVPVAEIEKLENQHFDEITKAGLK NGN
>sp|P0ABK2|CYDB_ECOLI Cytochrome d ubiquinol oxidase subunit 2 OS=Escherichia coli (strain K12) GN=cydB PE=1 SV=1
MIDYEVLRFIWWLLVGVLLIGFAVTDGFDMGVGMLTRFLGRNDTERRIMINSIAPHWDGN QVWLITAGGALFAAWPMVYAAAFSGFYVAMILVLASLFFRPVGFDYRSKIEETRWRNMWD WGIFIGSFVPPLVIGVAFGNLLQGVPFNVDEYLRLYYTGNFFQLLNPFGLLAGVVSVGMI ITQGATYLQMRTVGELHLRTRATAQVAALVTLVCFALAGVWVMYGIDGYVVKSTMDHYAA SNPLNKEVVREAGAWLVNFNNTPILWAIPALGVVLPLLTILTARMDKAAWAFVFSSLTLA CIILTAGIAMFPFVMPSSTMMNASLTMWDATSSQLTLNVMTWVAVVLVPIILLYTAWCYW KMFGRITKEDIERNTHSLY
>sp|Q6MIR4|NUOB_BDEBA NADH-quinone oxidoreductase subunit B OS=Bdellovibrio bacteriovorus (strain ATCC 15356 / DSM 50701 / NCIB 9529 / HD100) GN=nuoB PE=3 SV=1
MHNEQVQGLVSHDGMTGTQAVDDMSRGFAFTSKLDAIVAWGRKNSLWPMPYGTACCGIEF MSVMGPKYDLARFGAEVARFSPRQADLLVVAGTITEKMAPVIVRIYQQMLEPKYVLSMGA CASSGGFYRAYHVLQGVDKVIPVDVYIPGCPPTPEAVMDGIMALQRMIATNQPRPWKDNW KSPYEQA
>sp|Q89AU5|NUOB_BUCBP NADH-quinone oxidoreductase subunit B OS=Buchnera aphidicola subsp. Baizongia pistaciae (strain Bp) GN=nuoB PE=3 SV=1
MKYTLTRVNISDDDQNYPREKKIQVSDPTKKYIQKNVFMGTLSKVLHNLVNWGRKNSLWP YNFGLSCCYVEMVTSFTSVHDISRFGSEVLRASPRQADFMVIAGTPFIKMVPIIQRLYDQ MLEPKWVISMGSCANSGGMYDIYSVVQGVDKFLPVDVYIPGCPPRPEAYIHGLMLLQKSI SKERRPLSWIIGEQGIYKANFNSEKKNLRKMRNLVKYSQDKN
>sp|Q82DY0|NUOB1_STRAW NADH-quinone oxidoreductase subunit B 1 OS=Streptomyces avermitilis (strain ATCC 31267 / DSM 46492 / JCM 5070 / NCIMB 12804 / NRRL 8165 / MA-4680) GN=nuoB1 PE=3 SV=1
MGLEEKLPSGFLLTTVEQAAGWVRKASVFPATFGLACCAIEMMTTGAGRYDLARFGMEVF RGSPRQADLMIVAGRVSQKMAPVLRQVYDQMPNPKWVISMGVCASSGGMFNNYAIVQGVD HIVPVDIYLPGCPPRPEMLIDAILKLHQKIQSSKLGVNAEEAAREAEEAALKALPTIEMK GLLR
Astonishingly, certain bacteria that "everyone knows" are anaerobic turned up as aerobic when checked with the above Blast-query. (For example: Bacteroides fragilis, Desulfovibrio gigas, Moorella thermoacetica, and others.) It seems quite a number of so-called anaerobes have non-copper (heme only) cytochrome oxidases. (See this paper for further discussion.)
In any event, my Blast search turned up 1,089 positives (putative aerobes, some facultatively anaerobic) out of 1,373 bacterial species. I tagged the non-positives as anaerobes.
Of the 284 putative anaerobes, 87 scored positive in a Blast protein search (t-blast-n) for catalase. I used the following catalase sequences in my query:
>sp|B0C4G1|KATG_ACAM1 Catalase-peroxidase OS=Acaryochloris marina (strain MBIC 11017) GN=katG PE=3 SV=1
MSSASKCPFSGGALKFTAGSGTANRDWWPNQLNLQILRQHSPKSNPMDKAFNYAEAFKSL DLADVKQDIFDLMKSSQDWWPADYGHYGPLFIRMAWHSAGTYRIGDGRGGAGTGNQRFAP INSWPDNANLDKARMLLWPIKQKYGAKISWADLMILAGNCALESMGFKTFGFAGGREDIW EPEEDIYWGAETEWLGDQRYTGDRDLEATLGAVQMGLIYVNPEGPNGHPDPVASGRDIRE TFGRMAMNDEETVALTAGGHTFGKCHGAGDDAHVGPEPEGARIEDQCLGWKSSFGTGKGV HAITSGIEGAWTTNPTQWDNNYFENLFGYEWELTKSPAGANQWVPQGGAGANTVPDAHDP SRRHAPIMTTADMAMRMDPIYSPISRRFLDNPDQFADAFARAWFKLTHRDMGPRSRYLGP EVPEEELIWQDPVPAVNHELINEQDIATLKSQILATNLTVSQLVSTAWASAVTYRNSDKR GGANGARIRLAPQRDWEVNQPAQLATVLQTLEAVQTTFNHSQIGGKRVSLADLIVLGGCA GVEQAAKNAGWYDVKVPFKPGRTDATQAQTDVTSFAVLEPRADGFRNYLKGHYPVSAEEL LVDKAQLLTLTAPEMTVLVGGLRVLNANVGQAQHGVFTHRPESLTNDFFLNLLDMSVTWA ATSEAEEVFEGRDRKTGALKWTGTRVDLIFGSNSQLRALAEVYGCEDSQQRFVQDFVAAW DKVMNLDRFDLA
>tr|D9RGS2|D9RGS2_STAAJ Catalase OS=Staphylococcus aureus (strain JKD6159) GN=katE PE=3 SV=1
MSQQDKKLTGVFGHPVSDRENSMTAGPRGPLLMQDIYFLEQMSQFDREVIPERRMHAKGS GAFGTFTVTKDITKYTNAKIFSEIGKQTEMFARFSTVAGERGAADAERDIRGFALKFYTE EGNWDLVGNNTPVFFFRDPKLFVSLNRAVKRDPRTNMRDAQNNWDFWTGLPEALHQVTIL MSDRGIPKDLRHMHGFGSHTYSMYNDSGERVWVKFHFRTQQGIENLTDEEAAEIIASDRD SSQRDLFEAIEKGDYPKWTMYIQVMTEEQAKSHKDNPFDLTKVWYHDEYPLIEVGEFELN RNPDNYFMDVEQAAFAPTNIIPGLDFSPDKMLQGRLFSYGDAQRYRLGVNHWQIPVNQPK GVGIENICPFSRDGQMRVVDNNQGGGTHYYPNNHGKFDSQPEYKKPPFPTDGYGYEYNQR QDDDNYFEQPGKLFRLQSEDAKERIFTNTANAMEGVTDDVKRRHIRHCYKADPEYGKGVA KALGIDINSIDLETENDETYENFEK
>sp|P60355|MCAT_LACPN Manganese catalase OS=Lactobacillus plantarum PE=1 SV=1
MFKHTRKLQYNAKPDRSDPIMARRLQESLGGQWGETTGMMSYLSQGWASTGAEKYKDLLL DTGTEEMAHVEMISTMIGYLLEDAPFGPEDLKRDPSLATTMAGMDPEHSLVHGLNASLNN PNGAAWNAGYVTSSGNLVADMRFNVVRESEARLQVSRLYSMTEDEGVRDMLKFLLARETQ HQLQFMKAQEELEEKYGIIVPGDMKEIEHSEFSHVLMNFSDGDGSKAFEGQVAKDGEKFT YQENPEAMGGIPHIKPGDPRLHNHQG
>sp|P42321|CATA_PROMI Catalase OS=Proteus mirabilis GN=katA PE=1 SV=1
MEKKKLTTAAGAPVVDNNNVITAGPRGPMLLQDVWFLEKLAHFDREVIPERRMHAKGSGA FGTFTVTHDITKYTRAKIFSEVGKKTEMFARFSTVAGERGAADAERDIRGFALKFYTEEG NWDMVGNNTPVFYLRDPLKFPDLNHIVKRDPRTNMRNMAYKWDFFSHLPESLHQLTIDMS DRGLPLSYRFVHGFGSHTYSFINKDNERFWVKFHFRCQQGIKNLMDDEAEALVGKDRESS QRDLFEAIERGDYPRWKLQIQIMPEKEASTVPYNPFDLTKVWPHADYPLMDVGYFELNRN PDNYFSDVEQAAFSPANIVPGISFSPDKMLQGRLFSYGDAHRYRLGVNHHQIPVNAPKCP FHNYHRDGAMRVDGNSGNGITYEPNSGGVFQEQPDFKEPPLSIEGAADHWNHREDEDYFS QPRALYELLSDDEHQRMFARIAGELSQASKETQQRQIDLFTKVHPEYGAGVEKAIKVLEG KDAK
>sp|Q9Z598|CATA_STRCO Catalase OS=Streptomyces coelicolor (strain ATCC BAA-471 / A3(2) / M145) GN=katA PE=3 SV=1
MSQRVLTTESGAPVADNQNSASAGIGGPLLIQDQHLIEKLARFNRERIPERVVHARGSGA YGHFEVTDDVSGFTHADFLNTVGKRTEVFLRFSTVADSLGGADAVRDPRGFALKFYTEEG NYDLVGNNTPVFFIKDPIKFPDFIHSQKRDPFTGRQEPDNVFDFWAHSPEATHQITWLMG DRGIPASYRHMDGFGSHTYQWTNARGESFFVKYHFKTDQGIRCLTADEAAKLAGEDPTSH QTDLVQAIERGVYPSWTLHVQLMPVAEAANYRFNPFDVTKVWPHADYPLKRVGRLVLDRN PDNVFAEVEQAAFSPNNFVPGIGPSPDKMLQGRLFAYADAHRYRLGVNHTQLAVNAPKAV PGGAANYGRDGLMAANPQGRYAKNYEPNSYDGPAETGTPLAAPLAVSGHTGTHEAPLHTK DDHFVQAGALYRLMSEDEKQRLVANLAGGLSQVSRNDVVEKNLAHFHAADPEYGKRVEEA VRALRED
>Haloarcula marismortui strain ATCC 43049(v1, unmasked), Name: YP_136584.1, katG1, rrnAC2018, Type: CDS, Feature Location: (Chr: I, complement(1808213..1810405)) Genomic Location: 1808213-1810405
MLKTVLMPSPSKCSLMAKRDQDWSPNQLRLDILDQNARDADPRGTGFDYAEEFQELDLDAVKADLEELMTSSQDWWPADYGHYGPLFIRMAWHSAGTYRTTDGRGGASGGRQRFAPLNSWPDNANLDKARRLLWPIKKKYGRKLSWADLIVLAGNHAIESMGLKTFGWAGGREDAFEPDEAVDWGPEDEMEAHQSERRTDDGELKEPLGAAVMGLIYVDPEGPNGNPDPLASAENIRESFGRMAMNDEETAALIAGGHTFGKVHGADDPEENLGDVPEDAPIEQMGLGWENDYGSGKAGDTITSGIEGPWTQAPIEWDNGYIDNLLDYEWEPEKGPGGAWQWTPTDEALANTVPDAHDPSEKQTPMMLTTDIALKRDPDYREVMERFQENPMEFGINFARAWYKLIHRDMGPPERFLGPDAPDEEMIWQDPVPDVDHDLIGDEEVAELKTDILETDLTVSQLVKTAWASASTYRDSDKRGGANGARIRLEPQKNWEVNEPAQLETVLATLEEIQAEFNSARTDDTRVSLADLIVLGGNAAVEQAAADAGYDVTVPFEPGRTDATPEQTDVDSFEALKPRADGFRNYARDDVDVPAEELLVDRADLLDLTPEEMTVLVGGLRSLGATYQDSDLGVFTDEPGTLTNDFFEVVLGMDTEWEPVSESKDVFEGYDRETGEQTWAASRVDLIFGSHSRLRAIAEVYGADGAEAELVDDFVDAWHKVMRLDRFDLE
>sp|B2TJE9|KATG_CLOBB Catalase-peroxidase OS=Clostridium botulinum (strain Eklund 17B / Type B) GN=katG PE=3 SV=1
MTENKCPVTGKMGKATAGSGTTNKDWWPNQLNLNILHQNSQLSNPMSKDFNYAEEFKKLD FQALKVDLYMLMTDSQIWWPADYGNYGPLFIRMAWHSAGTYRVGDGRGGGSLGLQRFAPL NSWPDNINLDKARRLLWPIKKKYGNKISWADLLILTGNCALESMGLKTLGFGGGRVDVWE PQEDIYWGSEKEWLGDEREKGDKELENPLAAVQMGLIYVNPEGPNGNPDPLGSAHDVRET FARMAMNDEETVALIAGGHTFGKCHGAASPSYVGPAPEAAPIEEQGLGWKNTYGSGNGDD TIGSGLEGAWKANPTKWTMGYLKTLFKYDWELVKSPAGAYQWLAKNVDEEDMVIDAEDST KKHRPMMTTADLGLRYDPIYEPIARNYLKNPEKFAHDFASAWFKLTHRDMGPISRYLGPE VPKESFIWQDPIPLVKHKLITKKDITHIKKKILDSGLSISDLVATAWASASTFRGSDKRG GANGGRIRLEPQKNWEVNEPKKLNNVLNTLKQIKENFNSSHSKDKKVSLADIIILGGCVG IEQAAKRAGYNINVPFIPGRTDAIQEQTDVKSFAVLEPKEDGFRNYLKTKYVVKPEDMLI DRAQLLTLTAPEMTVLIGGMRVLNCNYNKSKDGVFTNRPECLTNDFFVNLLDMNTVWKPK SEDKDRFEGFDRETGELKWTATRVDLIFGSNSQLRAIAEVYACDDNKEKFIQDFIFAWNK IMNADRFEIK
>sp|Q59635|CATB_PSEAE Catalase OS=Pseudomonas aeruginosa (strain ATCC 15692 / PAO1 / 1C / PRS 101 / LMG 12228) GN=katB PE=3 SV=1
MNPSLNAFRPGRLLVAASLTASLLSLSVQAATLTRDNGAPVGDNQNSQTAGPNGSVLLQD VQLLQKLQRFDRERIPERVVHARGTGAHGEFVASADISDLSMAKVFRKGEKTPVFVRFSA VVHGNHSPETLRDPRGFATKFYTADGNWDLVGNNFPTFFIRDAIKFPDMVHAFKPDPRSN LDDDSRRFDFFSHVPEATRTLTLLYSNEGTPASYREMDGNSVHAYKLVNARGEVHYVKFH WKSLQGQKNLDPKQVAEVQGRDYSHMTNDLVSAIRKGDFPKWDLYIQVLKPEDLAKFDFD PLDATKIWPGIPERKIGQMVLNRNVDNFFQETEQVAMAPSNLVPGIEPSEDRLLQGRLFA YADTQMYRVGANGLGLPVNRPRSEVNTVNQDGALNAGHSTSGVNYQPSRLDPREEQASAR YVRTPLSGTTQQAKIQREQNFKQTGELFRSYGKKDQADLIASLGGALAITDDESKYIMLS YFYKADSDYGTGLAKVAGADLQRVRQLAAKLQD
The first of these is a cyanobacterial katG (large subunit) type of catalase, perhaps representative of primitive protobacterial catalase. The second sequence in the above list is classic Staphylococcus catalase (katE). The third is a manganese-containing catalase from Lactobacillus. (This brought the most hits, by the way.) The others are, in turn, katA catalase from Proteus and Streptomyces, two organisms that are far apart in genomic G+C content (and rather distant phylogenetically); an Archaeal catalase (even though none of the 1,373 species in my organism list was Archaeal in origin; but you never know whether a given bacterium may have obtained its catalase through horizontal gene transfer); then a known-valid anaerobic catalase from Clostridium botulinum, and finally a Pseudomonas katB catalase. The idea was to cover as much ground, phylogenetically and enzymatically, as possible, with big and small-subunit catalases, of the heme as well as the manganese variety, from aerobic and anaerobic bacteria of high and low genomic G+C content, as well as an archaeal catalase for good measure.
Here, then, finally, is the list of 87 catalase-positive strict anaerobes:
Note that these are all bacteria; no archaeons are included. (And yes, there are catalase-positive anaerobes among the Archaea.) The reason you don't see Bacteroides fragilis (which is catalase-positive) on the list is that, as explained before, B. fragilis ended up being classified an aerobe by my cytochrome-oxidase-based initial search. Even though "everybody knows" B. fragilis is anaerobic.
Incidentally, Blast searches were done with an E-value cutoff of 1e-5, to reduce the chance of false positives. (E-value is a measure of how likely it is that a given Blast match could have occurred due to chance. A threshold value of 1e-5 means the only matches that will be accepted are those that have less than a 1-in-100,000 chance of occurring by chance.)
If you learn of any other catalase-positive anaerobes that should be on this list, do be sure to let me know!
I've written on this subject before, so I won't bore you with a proper debunking of all aspects of the catalase myth here. (For that, see this post.) Right now, I just want to emphasize one point, which is that, contrary to myth, quite a few strict anaerobes do have catalase. I've listed 87 examples by name below. (Scroll down.)
I have to admit, even I was shocked to find there are 87 species of catalase-positive strict anaerobes among the eubacteria. It's about quadruple the number I would have expected.
If you're curious how I came up with a list of 87 catalase-positive anaerobes, here's how. First, I assembled a sizable (N=1373) list of bacteria, unduplicated at the species level. (So in other words, E. coli is listed only once, Staphylococcus aureus is listed only once, etc. No species is listed twice.) I then used the free/online CoGeBlast tool to run two Blast searches: one designed to identify aerobes, and another to identify catalase-positive organisms. In the end, I had all 1,373 organisms tagged as to whether each was aerobic, anaerobic, catalase-positive, or catalase-negative.
It's not as easy as you'd think to identify strict anaerobes. There is no single enzymatic marker that can be used to identify anaerobes reliably (across 1,373 species), as far as I know. I took the opposite approach, tagging as aerobic any organism that produces cytochrome c oxidase and/or NADH dehydrogenase. (These are enzymes involved in classic oxidative phosphorylation of the kind no strict anaerobe participates in.) In particular, I used the following set of amino acid sequences as markers of aerobic respiration (non-biogeeks, scroll down):
>sp|Q6MIR4|NUOB_BDEBA NADH-quinone oxidoreductase subunit B OS=Bdellovibrio bacteriovorus (strain ATCC 15356 / DSM 50701 / NCIB 9529 / HD100) GN=nuoB PE=3 SV=1
MHNEQVQGLVSHDGMTGTQAVDDMSRGFAFTSKLDAIVAWGRKNSLWPMPYGTACCGIEF MSVMGPKYDLARFGAEVARFSPRQADLLVVAGTITEKMAPVIVRIYQQMLEPKYVLSMGA CASSGGFYRAYHVLQGVDKVIPVDVYIPGCPPTPEAVMDGIMALQRMIATNQPRPWKDNW KSPYEQA
>sp|P0ABJ3|CYOC_ECOLI Cytochrome o ubiquinol oxidase subunit 3 OS=Escherichia coli (strain K12) GN=cyoC PE=1 SV=1
MATDTLTHATAHAHEHGHHDAGGTKIFGFWIYLMSDCILFSILFATYAVLVNGTAGGPTG KDIFELPFVLVETFLLLFSSITYGMAAIAMYKNNKSQVISWLALTWLFGAGFIGMEIYEF HHLIVNGMGPDRSGFLSAFFALVGTHGLHVTSGLIWMAVLMVQIARRGLTSTNRTRIMCL SLFWHFLDVVWICVFTVVYLMGAM
>sp|Q9I425|CYOC_PSEAE Cytochrome o ubiquinol oxidase subunit 3 OS=Pseudomonas aeruginosa (strain ATCC 15692 / PAO1 / 1C / PRS 101 / LMG 12228) GN=cyoC PE=3 SV=1
MSTAVLNKHLADAHEVGHDHDHAHDSGGNTVFGFWLYLMTDCVLFASVFATYAVLVHHTA GGPSGKDIFELPYVLVETAILLVSSCTYGLAMLSAHKGAKGQAIAWLGVTFLLGAAFIGM EINEFHHLIAEGFGPSRSAFLSSFFTLVGMHGLHVSAGLLWMLVLMAQIWTRGLTAQNNT RMMCLSLFWHFLDIVWICVFTVVYLMGAL
>tr|Q7VDD9|Q7VDD9_PROMA Cytochrome c oxidase subunit III OS=Prochlorococcus marinus (strain SARG / CCMP1375 / SS120) GN=cyoC PE=3 SV=1
MTTISSVDKKAEELTSQTEEHPDLRLFGLVSFLVADGMTFAGFFAAYLTFKAVNPLLPDA IYELELPLPTLNTILLLVSSATFHRAGKALEAKESEKCQRWLLITAGLGIAFLVSQMFEY FTLPFGLTDNLYASTFYALTGFHGLHVTLGAIMILIVWWQARSPGGRITTENKFPLEAAE LYWHFVDGIWVILFIILYLL
>sp|Q8KS19|CCOP2_PSEST Cbb3-type cytochrome c oxidase subunit CcoP2 OS=Pseudomonas stutzeri GN=ccoP2 PE=1 SV=1
MTSFWSWYVTLLSLGTIAALVWLLLATRKGQRPDSTEETVGHSYDGIEEYDNPLPRWWFM LFVGTVIFALGYLVLYPGLGNWKGILPGYEGGWTQVKEWQREMDKANEQYGPLYAKYAAM PVEEVAKDPQALKMGGRLFASNCSVCHGSDAKGAYGFPNLTDDDWLWGGEPETIKTTILH GRQAVMPGWKDVIGEEGIRNVAGYVRSLSGRDTPEGISVDIEQGQKIFAANCVVCHGPEA KGVTAMGAPNLTDNVWLYGSSFAQIQQTLRYGRNGRMPAQEAILGNDKVHLLAAYVYSLS QQPEQ
>sp|P57542|CYOC_BUCAI Cytochrome o ubiquinol oxidase subunit 3 OS=Buchnera aphidicola subsp. Acyrthosiphon pisum (strain APS) GN=cyoC PE=3 SV=1
MIENKFNNTILNSNSSTHDKISETKKLFGLWIYLMSDCIMFAVLFAVYAIVSSNISINLI SNKIFNLSSILLETFLLLLSSLSCGFVVIAMNQKRIKMIYSFLTITFIFGLIFLLMEVHE FYELIIENFGPDKNAFFSIFFTLVATHGVHIFFGLILILSILYQIKKLGLTNSIRTRILC FSVFWHFLDIIWICVFTFVYLNGAI
>sp|O24958|CCOP_HELPY Cbb3-type cytochrome c oxidase subunit CcoP OS=Helicobacter pylori (strain ATCC 700392 / 26695) GN=ccoP PE=3 SV=1
MDFLNDHINVFGLIAALVILVLTIYESSSLIKEMRDSKSQGELVENGHLIDGIGEFANNV PVGWIASFMCTIVWAFWYFFFGYPLNSFSQIGQYNEEVKAHNQKFEAKWKHLGQKELVDM GQGIFLVHCSQCHGITAEGLHGSAQNLVRWGKEEGIMDTIKHGSKGMDYLAGEMPAMELD EKDAKAIASYVMAELSSVKKTKNPQLIDKGKELFESMGCTGCHGNDGKGLQENQVFAADL TAYGTENFLRNILTHGKKGNIGHMPSFKYKNFSDLQVKALLNLSNR
>sp|P0ABI8|CYOB_ECOLI Ubiquinol oxidase subunit 1 OS=Escherichia coli (strain K12) GN=cyoB PE=1 SV=1
MFGKLSLDAVPFHEPIVMVTIAGIILGGLALVGLITYFGKWTYLWKEWLTSVDHKRLGIM YIIVAIVMLLRGFADAIMMRSQQALASAGEAGFLPPHHYDQIFTAHGVIMIFFVAMPFVI GLMNLVVPLQIGARDVAFPFLNNLSFWFTVVGVILVNVSLGVGEFAQTGWLAYPPLSGIE YSPGVGVDYWIWSLQLSGIGTTLTGINFFVTILKMRAPGMTMFKMPVFTWASLCANVLII ASFPILTVTVALLTLDRYLGTHFFTNDMGGNMMMYINLIWAWGHPEVYILILPVFGVFSE IAATFSRKRLFGYTSLVWATVCITVLSFIVWLHHFFTMGAGANVNAFFGITTMIIAIPTG VKIFNWLFTMYQGRIVFHSAMLWTIGFIVTFSVGGMTGVLLAVPGADFVLHNSLFLIAHF HNVIIGGVVFGCFAGMTYWWPKAFGFKLNETWGKRAFWFWIIGFFVAFMPLYALGFMGMT RRLSQQIDPQFHTMLMIAASGAVLIALGILCLVIQMYVSIRDRDQNRDLTGDPWGGRTLE WATSSPPPFYNFAVVPHVHERDAFWEMKEKGEAYKKPDHYEEIHMPKNSGAGIVIAAFST IFGFAMIWHIWWLAIVGFAGMIITWIVKSFDEDVDYYVPVAEIEKLENQHFDEITKAGLK NGN
>sp|P0ABK2|CYDB_ECOLI Cytochrome d ubiquinol oxidase subunit 2 OS=Escherichia coli (strain K12) GN=cydB PE=1 SV=1
MIDYEVLRFIWWLLVGVLLIGFAVTDGFDMGVGMLTRFLGRNDTERRIMINSIAPHWDGN QVWLITAGGALFAAWPMVYAAAFSGFYVAMILVLASLFFRPVGFDYRSKIEETRWRNMWD WGIFIGSFVPPLVIGVAFGNLLQGVPFNVDEYLRLYYTGNFFQLLNPFGLLAGVVSVGMI ITQGATYLQMRTVGELHLRTRATAQVAALVTLVCFALAGVWVMYGIDGYVVKSTMDHYAA SNPLNKEVVREAGAWLVNFNNTPILWAIPALGVVLPLLTILTARMDKAAWAFVFSSLTLA CIILTAGIAMFPFVMPSSTMMNASLTMWDATSSQLTLNVMTWVAVVLVPIILLYTAWCYW KMFGRITKEDIERNTHSLY
>sp|Q6MIR4|NUOB_BDEBA NADH-quinone oxidoreductase subunit B OS=Bdellovibrio bacteriovorus (strain ATCC 15356 / DSM 50701 / NCIB 9529 / HD100) GN=nuoB PE=3 SV=1
MHNEQVQGLVSHDGMTGTQAVDDMSRGFAFTSKLDAIVAWGRKNSLWPMPYGTACCGIEF MSVMGPKYDLARFGAEVARFSPRQADLLVVAGTITEKMAPVIVRIYQQMLEPKYVLSMGA CASSGGFYRAYHVLQGVDKVIPVDVYIPGCPPTPEAVMDGIMALQRMIATNQPRPWKDNW KSPYEQA
>sp|Q89AU5|NUOB_BUCBP NADH-quinone oxidoreductase subunit B OS=Buchnera aphidicola subsp. Baizongia pistaciae (strain Bp) GN=nuoB PE=3 SV=1
MKYTLTRVNISDDDQNYPREKKIQVSDPTKKYIQKNVFMGTLSKVLHNLVNWGRKNSLWP YNFGLSCCYVEMVTSFTSVHDISRFGSEVLRASPRQADFMVIAGTPFIKMVPIIQRLYDQ MLEPKWVISMGSCANSGGMYDIYSVVQGVDKFLPVDVYIPGCPPRPEAYIHGLMLLQKSI SKERRPLSWIIGEQGIYKANFNSEKKNLRKMRNLVKYSQDKN
>sp|Q82DY0|NUOB1_STRAW NADH-quinone oxidoreductase subunit B 1 OS=Streptomyces avermitilis (strain ATCC 31267 / DSM 46492 / JCM 5070 / NCIMB 12804 / NRRL 8165 / MA-4680) GN=nuoB1 PE=3 SV=1
MGLEEKLPSGFLLTTVEQAAGWVRKASVFPATFGLACCAIEMMTTGAGRYDLARFGMEVF RGSPRQADLMIVAGRVSQKMAPVLRQVYDQMPNPKWVISMGVCASSGGMFNNYAIVQGVD HIVPVDIYLPGCPPRPEMLIDAILKLHQKIQSSKLGVNAEEAAREAEEAALKALPTIEMK GLLR
Astonishingly, certain bacteria that "everyone knows" are anaerobic turned up as aerobic when checked with the above Blast-query. (For example: Bacteroides fragilis, Desulfovibrio gigas, Moorella thermoacetica, and others.) It seems quite a number of so-called anaerobes have non-copper (heme only) cytochrome oxidases. (See this paper for further discussion.)
In any event, my Blast search turned up 1,089 positives (putative aerobes, some facultatively anaerobic) out of 1,373 bacterial species. I tagged the non-positives as anaerobes.
Of the 284 putative anaerobes, 87 scored positive in a Blast protein search (t-blast-n) for catalase. I used the following catalase sequences in my query:
>sp|B0C4G1|KATG_ACAM1 Catalase-peroxidase OS=Acaryochloris marina (strain MBIC 11017) GN=katG PE=3 SV=1
MSSASKCPFSGGALKFTAGSGTANRDWWPNQLNLQILRQHSPKSNPMDKAFNYAEAFKSL DLADVKQDIFDLMKSSQDWWPADYGHYGPLFIRMAWHSAGTYRIGDGRGGAGTGNQRFAP INSWPDNANLDKARMLLWPIKQKYGAKISWADLMILAGNCALESMGFKTFGFAGGREDIW EPEEDIYWGAETEWLGDQRYTGDRDLEATLGAVQMGLIYVNPEGPNGHPDPVASGRDIRE TFGRMAMNDEETVALTAGGHTFGKCHGAGDDAHVGPEPEGARIEDQCLGWKSSFGTGKGV HAITSGIEGAWTTNPTQWDNNYFENLFGYEWELTKSPAGANQWVPQGGAGANTVPDAHDP SRRHAPIMTTADMAMRMDPIYSPISRRFLDNPDQFADAFARAWFKLTHRDMGPRSRYLGP EVPEEELIWQDPVPAVNHELINEQDIATLKSQILATNLTVSQLVSTAWASAVTYRNSDKR GGANGARIRLAPQRDWEVNQPAQLATVLQTLEAVQTTFNHSQIGGKRVSLADLIVLGGCA GVEQAAKNAGWYDVKVPFKPGRTDATQAQTDVTSFAVLEPRADGFRNYLKGHYPVSAEEL LVDKAQLLTLTAPEMTVLVGGLRVLNANVGQAQHGVFTHRPESLTNDFFLNLLDMSVTWA ATSEAEEVFEGRDRKTGALKWTGTRVDLIFGSNSQLRALAEVYGCEDSQQRFVQDFVAAW DKVMNLDRFDLA
>tr|D9RGS2|D9RGS2_STAAJ Catalase OS=Staphylococcus aureus (strain JKD6159) GN=katE PE=3 SV=1
MSQQDKKLTGVFGHPVSDRENSMTAGPRGPLLMQDIYFLEQMSQFDREVIPERRMHAKGS GAFGTFTVTKDITKYTNAKIFSEIGKQTEMFARFSTVAGERGAADAERDIRGFALKFYTE EGNWDLVGNNTPVFFFRDPKLFVSLNRAVKRDPRTNMRDAQNNWDFWTGLPEALHQVTIL MSDRGIPKDLRHMHGFGSHTYSMYNDSGERVWVKFHFRTQQGIENLTDEEAAEIIASDRD SSQRDLFEAIEKGDYPKWTMYIQVMTEEQAKSHKDNPFDLTKVWYHDEYPLIEVGEFELN RNPDNYFMDVEQAAFAPTNIIPGLDFSPDKMLQGRLFSYGDAQRYRLGVNHWQIPVNQPK GVGIENICPFSRDGQMRVVDNNQGGGTHYYPNNHGKFDSQPEYKKPPFPTDGYGYEYNQR QDDDNYFEQPGKLFRLQSEDAKERIFTNTANAMEGVTDDVKRRHIRHCYKADPEYGKGVA KALGIDINSIDLETENDETYENFEK
>sp|P60355|MCAT_LACPN Manganese catalase OS=Lactobacillus plantarum PE=1 SV=1
MFKHTRKLQYNAKPDRSDPIMARRLQESLGGQWGETTGMMSYLSQGWASTGAEKYKDLLL DTGTEEMAHVEMISTMIGYLLEDAPFGPEDLKRDPSLATTMAGMDPEHSLVHGLNASLNN PNGAAWNAGYVTSSGNLVADMRFNVVRESEARLQVSRLYSMTEDEGVRDMLKFLLARETQ HQLQFMKAQEELEEKYGIIVPGDMKEIEHSEFSHVLMNFSDGDGSKAFEGQVAKDGEKFT YQENPEAMGGIPHIKPGDPRLHNHQG
>sp|P42321|CATA_PROMI Catalase OS=Proteus mirabilis GN=katA PE=1 SV=1
MEKKKLTTAAGAPVVDNNNVITAGPRGPMLLQDVWFLEKLAHFDREVIPERRMHAKGSGA FGTFTVTHDITKYTRAKIFSEVGKKTEMFARFSTVAGERGAADAERDIRGFALKFYTEEG NWDMVGNNTPVFYLRDPLKFPDLNHIVKRDPRTNMRNMAYKWDFFSHLPESLHQLTIDMS DRGLPLSYRFVHGFGSHTYSFINKDNERFWVKFHFRCQQGIKNLMDDEAEALVGKDRESS QRDLFEAIERGDYPRWKLQIQIMPEKEASTVPYNPFDLTKVWPHADYPLMDVGYFELNRN PDNYFSDVEQAAFSPANIVPGISFSPDKMLQGRLFSYGDAHRYRLGVNHHQIPVNAPKCP FHNYHRDGAMRVDGNSGNGITYEPNSGGVFQEQPDFKEPPLSIEGAADHWNHREDEDYFS QPRALYELLSDDEHQRMFARIAGELSQASKETQQRQIDLFTKVHPEYGAGVEKAIKVLEG KDAK
>sp|Q9Z598|CATA_STRCO Catalase OS=Streptomyces coelicolor (strain ATCC BAA-471 / A3(2) / M145) GN=katA PE=3 SV=1
MSQRVLTTESGAPVADNQNSASAGIGGPLLIQDQHLIEKLARFNRERIPERVVHARGSGA YGHFEVTDDVSGFTHADFLNTVGKRTEVFLRFSTVADSLGGADAVRDPRGFALKFYTEEG NYDLVGNNTPVFFIKDPIKFPDFIHSQKRDPFTGRQEPDNVFDFWAHSPEATHQITWLMG DRGIPASYRHMDGFGSHTYQWTNARGESFFVKYHFKTDQGIRCLTADEAAKLAGEDPTSH QTDLVQAIERGVYPSWTLHVQLMPVAEAANYRFNPFDVTKVWPHADYPLKRVGRLVLDRN PDNVFAEVEQAAFSPNNFVPGIGPSPDKMLQGRLFAYADAHRYRLGVNHTQLAVNAPKAV PGGAANYGRDGLMAANPQGRYAKNYEPNSYDGPAETGTPLAAPLAVSGHTGTHEAPLHTK DDHFVQAGALYRLMSEDEKQRLVANLAGGLSQVSRNDVVEKNLAHFHAADPEYGKRVEEA VRALRED
>Haloarcula marismortui strain ATCC 43049(v1, unmasked), Name: YP_136584.1, katG1, rrnAC2018, Type: CDS, Feature Location: (Chr: I, complement(1808213..1810405)) Genomic Location: 1808213-1810405
MLKTVLMPSPSKCSLMAKRDQDWSPNQLRLDILDQNARDADPRGTGFDYAEEFQELDLDAVKADLEELMTSSQDWWPADYGHYGPLFIRMAWHSAGTYRTTDGRGGASGGRQRFAPLNSWPDNANLDKARRLLWPIKKKYGRKLSWADLIVLAGNHAIESMGLKTFGWAGGREDAFEPDEAVDWGPEDEMEAHQSERRTDDGELKEPLGAAVMGLIYVDPEGPNGNPDPLASAENIRESFGRMAMNDEETAALIAGGHTFGKVHGADDPEENLGDVPEDAPIEQMGLGWENDYGSGKAGDTITSGIEGPWTQAPIEWDNGYIDNLLDYEWEPEKGPGGAWQWTPTDEALANTVPDAHDPSEKQTPMMLTTDIALKRDPDYREVMERFQENPMEFGINFARAWYKLIHRDMGPPERFLGPDAPDEEMIWQDPVPDVDHDLIGDEEVAELKTDILETDLTVSQLVKTAWASASTYRDSDKRGGANGARIRLEPQKNWEVNEPAQLETVLATLEEIQAEFNSARTDDTRVSLADLIVLGGNAAVEQAAADAGYDVTVPFEPGRTDATPEQTDVDSFEALKPRADGFRNYARDDVDVPAEELLVDRADLLDLTPEEMTVLVGGLRSLGATYQDSDLGVFTDEPGTLTNDFFEVVLGMDTEWEPVSESKDVFEGYDRETGEQTWAASRVDLIFGSHSRLRAIAEVYGADGAEAELVDDFVDAWHKVMRLDRFDLE
>sp|B2TJE9|KATG_CLOBB Catalase-peroxidase OS=Clostridium botulinum (strain Eklund 17B / Type B) GN=katG PE=3 SV=1
MTENKCPVTGKMGKATAGSGTTNKDWWPNQLNLNILHQNSQLSNPMSKDFNYAEEFKKLD FQALKVDLYMLMTDSQIWWPADYGNYGPLFIRMAWHSAGTYRVGDGRGGGSLGLQRFAPL NSWPDNINLDKARRLLWPIKKKYGNKISWADLLILTGNCALESMGLKTLGFGGGRVDVWE PQEDIYWGSEKEWLGDEREKGDKELENPLAAVQMGLIYVNPEGPNGNPDPLGSAHDVRET FARMAMNDEETVALIAGGHTFGKCHGAASPSYVGPAPEAAPIEEQGLGWKNTYGSGNGDD TIGSGLEGAWKANPTKWTMGYLKTLFKYDWELVKSPAGAYQWLAKNVDEEDMVIDAEDST KKHRPMMTTADLGLRYDPIYEPIARNYLKNPEKFAHDFASAWFKLTHRDMGPISRYLGPE VPKESFIWQDPIPLVKHKLITKKDITHIKKKILDSGLSISDLVATAWASASTFRGSDKRG GANGGRIRLEPQKNWEVNEPKKLNNVLNTLKQIKENFNSSHSKDKKVSLADIIILGGCVG IEQAAKRAGYNINVPFIPGRTDAIQEQTDVKSFAVLEPKEDGFRNYLKTKYVVKPEDMLI DRAQLLTLTAPEMTVLIGGMRVLNCNYNKSKDGVFTNRPECLTNDFFVNLLDMNTVWKPK SEDKDRFEGFDRETGELKWTATRVDLIFGSNSQLRAIAEVYACDDNKEKFIQDFIFAWNK IMNADRFEIK
>sp|Q59635|CATB_PSEAE Catalase OS=Pseudomonas aeruginosa (strain ATCC 15692 / PAO1 / 1C / PRS 101 / LMG 12228) GN=katB PE=3 SV=1
MNPSLNAFRPGRLLVAASLTASLLSLSVQAATLTRDNGAPVGDNQNSQTAGPNGSVLLQD VQLLQKLQRFDRERIPERVVHARGTGAHGEFVASADISDLSMAKVFRKGEKTPVFVRFSA VVHGNHSPETLRDPRGFATKFYTADGNWDLVGNNFPTFFIRDAIKFPDMVHAFKPDPRSN LDDDSRRFDFFSHVPEATRTLTLLYSNEGTPASYREMDGNSVHAYKLVNARGEVHYVKFH WKSLQGQKNLDPKQVAEVQGRDYSHMTNDLVSAIRKGDFPKWDLYIQVLKPEDLAKFDFD PLDATKIWPGIPERKIGQMVLNRNVDNFFQETEQVAMAPSNLVPGIEPSEDRLLQGRLFA YADTQMYRVGANGLGLPVNRPRSEVNTVNQDGALNAGHSTSGVNYQPSRLDPREEQASAR YVRTPLSGTTQQAKIQREQNFKQTGELFRSYGKKDQADLIASLGGALAITDDESKYIMLS YFYKADSDYGTGLAKVAGADLQRVRQLAAKLQD
The first of these is a cyanobacterial katG (large subunit) type of catalase, perhaps representative of primitive protobacterial catalase. The second sequence in the above list is classic Staphylococcus catalase (katE). The third is a manganese-containing catalase from Lactobacillus. (This brought the most hits, by the way.) The others are, in turn, katA catalase from Proteus and Streptomyces, two organisms that are far apart in genomic G+C content (and rather distant phylogenetically); an Archaeal catalase (even though none of the 1,373 species in my organism list was Archaeal in origin; but you never know whether a given bacterium may have obtained its catalase through horizontal gene transfer); then a known-valid anaerobic catalase from Clostridium botulinum, and finally a Pseudomonas katB catalase. The idea was to cover as much ground, phylogenetically and enzymatically, as possible, with big and small-subunit catalases, of the heme as well as the manganese variety, from aerobic and anaerobic bacteria of high and low genomic G+C content, as well as an archaeal catalase for good measure.
Here, then, finally, is the list of 87 catalase-positive strict anaerobes:
Acetohalobium arabaticum strain DSM 5501
Alkaliphilus metalliredigens strain QYMF
Alkaliphilus oremlandii strain OhILAs
Anaerococcus prevotii strain ACS-065-V-Col13
Anaerococcus vaginalis strain ATCC 51170
Anaerofustis stercorihominis strain DSM 17244
Anaerostipes caccae strain DSM 14662
Anaerostipes sp. strain 3_2_56FAA
Anaerotruncus colihominis strain DSM 17241
Bacteroides capillosus strain ATCC 29799
Bacteroides pectinophilus strain ATCC 43243
Brachyspira hyodysenteriae strain ATCC 49526; WA1
Brachyspira intermedia strain PWS/A
Brachyspira pilosicoli strain 95/1000
Candidatus Arthromitus sp. SFB-mouse-Japan
Carnobacterium sp. strain 17-4
Clostridium acetobutylicum strain ATCC 824
Clostridium asparagiforme strain DSM 15981
Clostridium bartlettii strain DSM 16795
Clostridium bolteae strain ATCC BAA-613
Clostridium botulinum A2 strain Kyoto
Clostridium butyricum strain 5521
Clostridium cellulovorans strain 743B
Clostridium cf. saccharolyticum strain K10
Clostridium citroniae strain WAL-17108
Clostridium clostridioforme strain 2_1_49FAA
Clostridium difficile QCD-37x79
Clostridium hathewayi strain WAL-18680
Clostridium hylemonae strain DSM 15053
Clostridium kluyveri strain DSM 555
Clostridium lentocellum strain DSM 5427
Clostridium leptum strain DSM 753
Clostridium ljungdahlii strain ATCC 49587
Clostridium novyi strain NT
Clostridium ramosum strain DSM 1402
Clostridium saccharolyticum strain WM1
Clostridium scindens strain ATCC 35704
Clostridium spiroforme strain DSM 1552
Clostridium sporogenes strain ATCC 15579
Clostridium tetani strain Massachusetts substrain E88
Coprobacillus sp. strain 3_3_56FAA
Coprococcus comes strain ATCC 27758
Coprococcus sp. strain ART55/1
Dethiobacter alkaliphilus strain AHT 1
Dorea formicigenerans strain 4_6_53AFAA
Dorea longicatena strain DSM 13814
Erysipelotrichaceae bacterium strain 21_3
Eubacterium dolichum strain DSM 3991
Eubacterium eligens strain ATCC 27750
Eubacterium siraeum strain 70/3
Eubacterium ventriosum strain ATCC 27560
Flavonifractor plautii strain ATCC 29863
Halothermothrix orenii strain DSM 9562; H 168
Holdemania filiformis strain DSM 12042
Lachnospiraceae bacterium strain 1_1_57FAA
Lactobacillus curvatus strain CRL 705
Lactobacillus sakei subsp. sakei strain 23K
Mahella australiensis strain 50-1 BON
Natranaerobius thermophilus strain JW/NM-WN-LF
Oscillibacter valericigenes strain Sjm18-20
Parabacteroides distasonis strain ATCC 8503
Parabacteroides johnsonii strain DSM 18315
Parabacteroides sp. strain D13
Pediococcus acidilactici strain DSM 20284
Pediococcus pentosaceus strain ATCC 25745
Pelotomaculum thermopropionicum strain SI
Pseudoflavonifractor capillosus strain ATCC 29799
Pseudoramibacter alactolyticus strain ATCC 23263
Roseburia hominis strain A2-183
Roseburia intestinalis strain M50/1
Ruminococcaceae bacterium strain D16
Ruminococcus bromii strain L2-63
Ruminococcus obeum strain A2-162
Ruminococcus sp. strain 18P13
Ruminococcus torques strain L2-14
Sphaerochaeta pleomorpha strain Grapes
Spirochaeta coccoides strain DSM 17374
Spirochaeta sp. strain Buddy
Subdoligranulum sp. strain 4_3_54A2FAA
Tepidanaerobacter sp. strain Re1
Thermoanaerobacter brockii subsp. finnii strain Ako-1
Thermoanaerobacter ethanolicus strain CCSD1
Thermoanaerobacter pseudethanolicus strain 39E; ATCC 33223
Thermoanaerobacter sp. strain X514
Thermosediminibacter oceani strain DSM 16646
Treponema brennaborense strain DSM 12168
Turicibacter sanguinis strain PC909
Note that these are all bacteria; no archaeons are included. (And yes, there are catalase-positive anaerobes among the Archaea.) The reason you don't see Bacteroides fragilis (which is catalase-positive) on the list is that, as explained before, B. fragilis ended up being classified an aerobe by my cytochrome-oxidase-based initial search. Even though "everybody knows" B. fragilis is anaerobic.
Incidentally, Blast searches were done with an E-value cutoff of 1e-5, to reduce the chance of false positives. (E-value is a measure of how likely it is that a given Blast match could have occurred due to chance. A threshold value of 1e-5 means the only matches that will be accepted are those that have less than a 1-in-100,000 chance of occurring by chance.)
If you learn of any other catalase-positive anaerobes that should be on this list, do be sure to let me know!
Subscribe to:
Posts (Atom)